摘要
接到一个wgs项目,要帮助客户统计vcf文件中snp突变频率,比较两个样品的突变位点。这个工作在上一个项目中是手动处理的,当时参考序列短,突变位点少。这次经过比对后,发现了有个样品有上万个snp位点,肯定不能用手动处理的方式。因此,写了一个脚本来统计各个样品的突变频率。
需要统计的信息
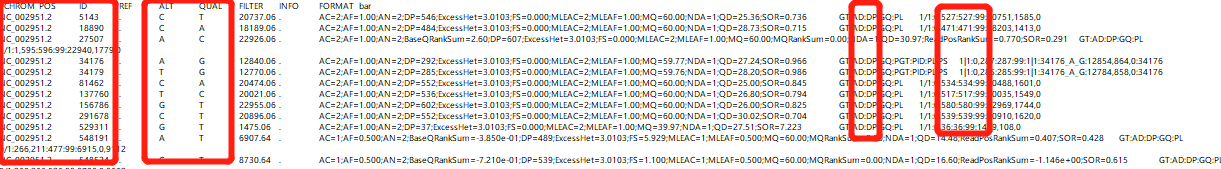
包括染色体,突变位置,参考位点,各样品突变位点,突变率(AD杂合位点覆盖度/DP总覆盖度)
环境与方法
python 3.7
R version 3.6.1
使用代码
统计各样品各位点突变频率
我们输入的vcf文件为GATK经过去重过滤后生成的,
bash python snp_frequency.py XX.snp.vcf
import csv
import sys
vcf_file = sys.argv[1] #文件调用vcf文件
genome_file = open(vcf_file,'r')
gtf_file = vcf_file.replace('_snp.vcf','')# 去除文件名后缀
genome_line = genome_file.readlines()
#genome_list = genome_line.split()
newgtf_name = '%s_stat'%gtf_file #保留样品名
desktop_path = './'
file_path = desktop_path+newgtf_name+'.csv' #确定文件路径和文件名,生成文件
file = open(file_path,'w')
for i in genome_line:
snp_pos = i.split('\t')
if "#" in snp_pos[0] and len(snp_pos) > 8:
title = snp_pos[0]+'\t'+snp_pos[1]+'\t'+snp_pos[3]+'\t'+snp_pos[4]+'\t'+'%s_FRE'%vcf_file+'\n'
file.write(title)
if "#" not in snp_pos[0] and len(snp_pos) > 8:
info = snp_pos[9].split(':')
AD = info[1].split(',')
AD_0 = int(AD[0])#转化为数字,后面需要加和
AD_1 = int(AD[1])
AD_total = int(AD[0])+int(AD[1]) #杂合子覆盖度求和
DP = info[2]
out_line = snp_pos[0]+'\t'+snp_pos[1]+'\t'+snp_pos[3]+'\t'+snp_pos[4]+'\t'+'%.0f'%AD_total+'/'+DP+'\n' #提取信息列
#print(out_line)
file.write(out_line)
file.close()
genome_file.close()
将比较样品进行合并,比较不同样品的突变情况。
WT=read.csv("USA_300_WT_stat.csv",header=T,sep='\t',check.names=F) S1=read.csv("USA_300_S1_stat.csv",header=T,sep='\t',check.names=F)
S2=read.csv("USA_300_S2_stat.csv",header=T,sep='\t',check.names=F)
WT_1 <- merge(WT,S1, all = TRUE) #合并WT和S1样品突变位点信息,不会对已有列进行重复(chr、pos、REF、ALT等)
WT_2 <- merge(WT,S2, all = TRUE) #同上
write.csv(WT_2,"USA_WT_vs_S2_snpstat.csv")
write.csv(WT_1,"USA_WT_vs_S1_snpstat.csv")
分析结果
得到.csv格式文件,这里用editplus打开

总结
WGS的最初的目的是为了寻找突变位点,这里是进行样品间比较,客户更需要的数据不是都发生突变的位点,而是某一个位点在其中一个样品发生突变(比如S1有S2无,反之亦然)。这样可以找到不同处理下的突变位点,深入研究。

版权声明:本文为yangl7原创文章,遵循CC 4.0 BY-SA版权协议,转载请附上原文出处链接和本声明。