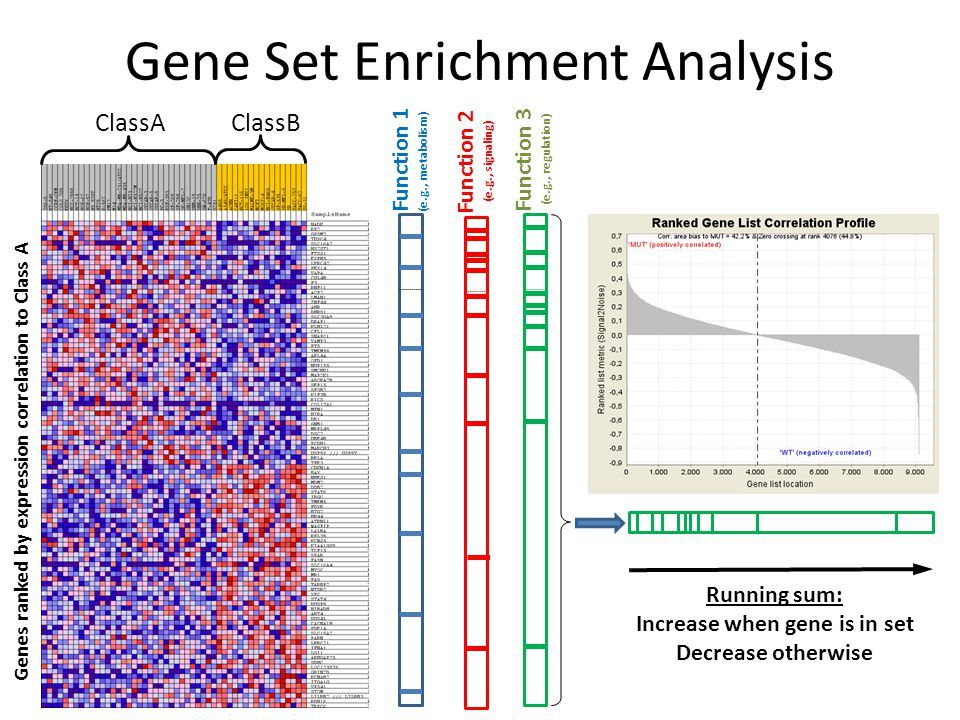
Gene Set Enrichment Analysis 基因富集分析
哈罗大家好!ヾ(≧▽≦*)o
年初在和老板研究 Identifying Cell Subpopulations 有关的课题,发现在发掘出 Characteristic Genes之后,有必要对这些基因做富集分析,来检测算法是否在正确的基因上对细胞进行分类。
奈何本程序猿没有系统学过生物有关的知识orz,因此只能慢慢整理和富集分析有关的知识,写成一份总结啦哈哈~这篇文章总结了一些博客和资料上有关基因富集分析的内容,大约描述了这是一个怎样的问题以及介绍了本地和在线富集分析的方式,如有错误内容,欢迎指正,谢谢,侵删。╮(╯▽╰)╭
目录
Gene Set Enrichment Analysis 基因富集分析
- 基因富集分析基本概念
- 基因富集分析基本流程
- 基因注释数据库 Gene Anotation Databas
- 京都基因与基因组百科全书 KEGG
- 基本概念
- 特点
- 基因本体数据库 GO
- 基本概念
- GO语义分类
- 注释结构
- GO Term
- 京都基因与基因组百科全书 KEGG
- 基因富集分析方法
- 方法分类
- 富集分析的冗余性问题
- GO富集分析
- 基本概念
- 分析原理
- GO富集分析的结果
- GO在线富集分析工具
- GO本地富集分析工具(GO::TermFinder)
- 参考资料
基因富集分析基本概念
基因富集分析是分析基因表达信息的一种方法。
富集是指将基因按照先验知识,也就是基因组注释信息,对基因进行分类的过程。基因经过分类后,能够帮助认知寻找到的基因是否具有某方面的共性(如功能、组成等等).
基因富集分析基本流程
1.首先通过生物学实验或计算机算法等手段,获取到一组基因,该组基因以基因列表、表达图谱、基因芯片等形式展示。
2.预先构建好基因注释数据库,如:GO、KEGG、MSigDB等。
3.通过特定的算法,根据基因注释数据库的知识, 对基因进行分类。
4.经过聚类后,去除冗余的结果,得到最终的基因富集结果。
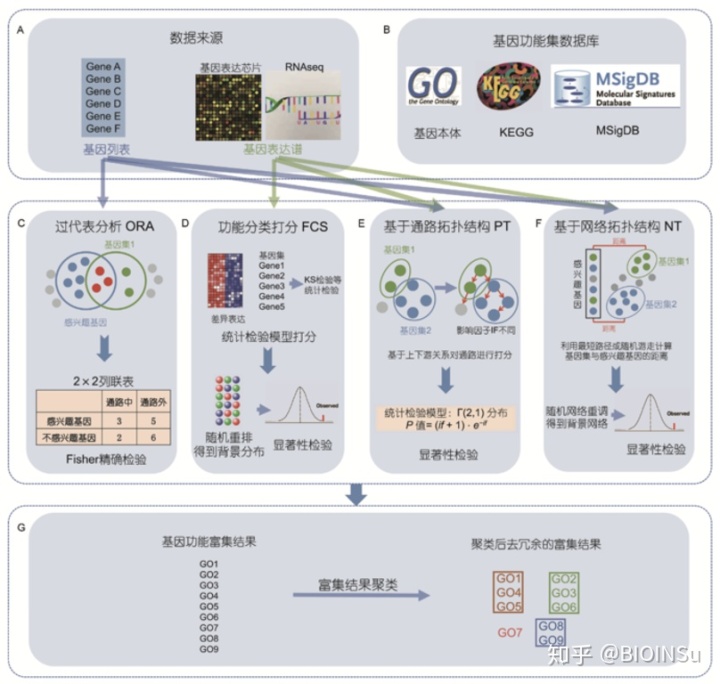
基因注释数据库 Gene Anotation Database
功能基因组学的任务之一,是进行基因组功能注释 (genome annotation),通过功能注释能够帮助了解基因功能。认识基因与疾病之间的关系,掌握基因的产物和基因在声明活动中的作用等等。
在基因组范围内描述蛋白质的功能十分复杂,最好的方法就是借助计算机程序,提供结构化的生物学模型,便于程序进行分析。
因此,不同的基因注释数据库被构建出来,从不同的生物学模型的角度对基因的功能进行注释存储。
基因注释数据库-京都基因与基因组百科全书 KEGG

基本概念
京都基因与基因组百科全书 (Kyoto encyclopedia of genes and genomes, KEGG),是系统分析基因功能与基因组信息的数据库,它整合了基因组学、生物化学和系统功能组学的信息,有助于研究者把基因及表达信息的过程作为一个网络进行整体研究。
特点
KEGG主要的特点是将基因与各种生化反应联系在了一起。它提供的整合代谢途径查询十分出色,还提供基于Java的图形工具访问基因组图谱,提供其他序列比较、图形比较和通路计算的工具。KEGG目前共包含了19个子数据库,他们被分类为系统信息、基因组信息和化学信息三个类别。
基因注释数据库-基因本体数据库 GO

基本概念
基因本体数据库 (Gene Ontology) 是GO组织 (Gene Ontology Consortium) 在2000年构建的一个结构化的标准生物模型,旨在建立基因及其产物知识的标准词汇体系,涵盖了基因的细胞组分 (cellular component)、分子功能 (molecular function)、生物学过程(biological process)。Term是GO里面的基本描述单元。
GO数据库官网如下:
http:// geneontology.org/
GO数据库最初收录的基因信息来三个生物数据库:果蝇、酵母、小鼠,随后从其他数据库相继收录植物、动物、微生物等更多的数据。GO术语在多个合作数据库中的统一使用,促进了各类数据库对基因描述的一致性。
GO语义分类
- 分子功能(Molecular Function)
描述在个体分子生物学上的活性,如催化活性或结合活性。
- 生物学过程(Biological Process)
由分子功能有序地组成的,具有多个步骤的一个过程。
- 细胞组件(Cellular Component)
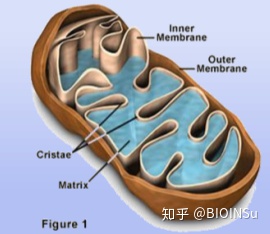
指基因产物位于何种细胞器或基因产物组中(如糙面内质网,核糖体,蛋白酶体等),即基因产物在什么地方起作用。
注释结构
GO注释体系的特点:GO通过控制注释词汇的层次结构 ,使得研究人员能够从不同层面查询和使用基因注释信息。从整体上看,GO注释系统是一个有向无环图 (Directed Acyclic Graphs),包含三个分支(即细胞组分、分子功能 、生物学过程)。注释系统中每一个节点都是基因或蛋白的一种描述,每一个节点就代表了一个基本描述单元 (Term) ,节点之间保持严格的关系。
注释结构示意图:
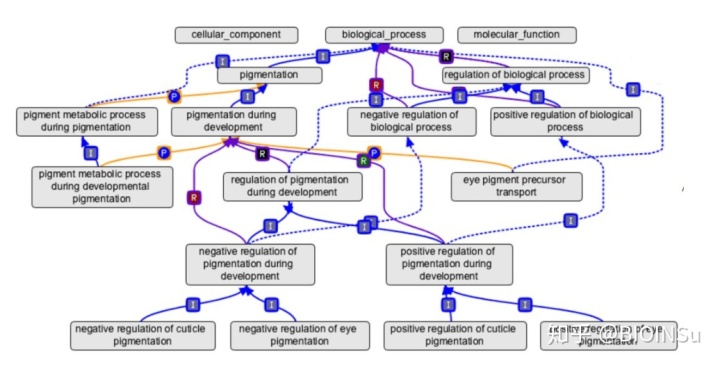
不同GO term之间的关系是多种多样的,以下只讨论4种常见的关系:
1.is a
is a关系的示意图如下
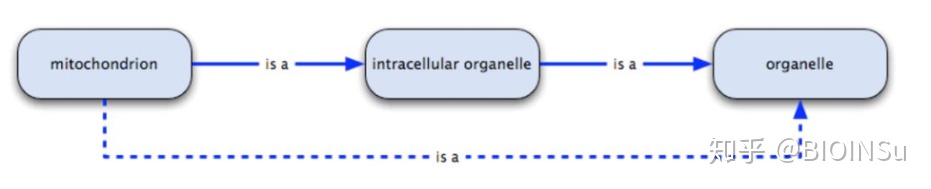
线粒体是细胞器的一种, 两者之间的关系是 线粒体is a细胞器。
2.part of
part of关系的示意图如下
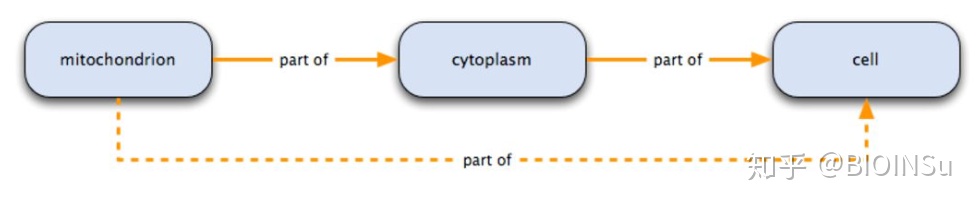
线粒体是细胞质的组成部分,二者之间的关系是线粒体part of细胞质。
3.hast part
has part关系的示意图如下
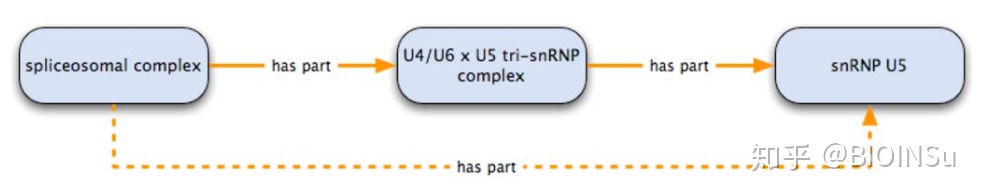
通常用于描述细胞内各种复合体之间的关系。
4.regulation
regulation 表示调控关系,示意图如下
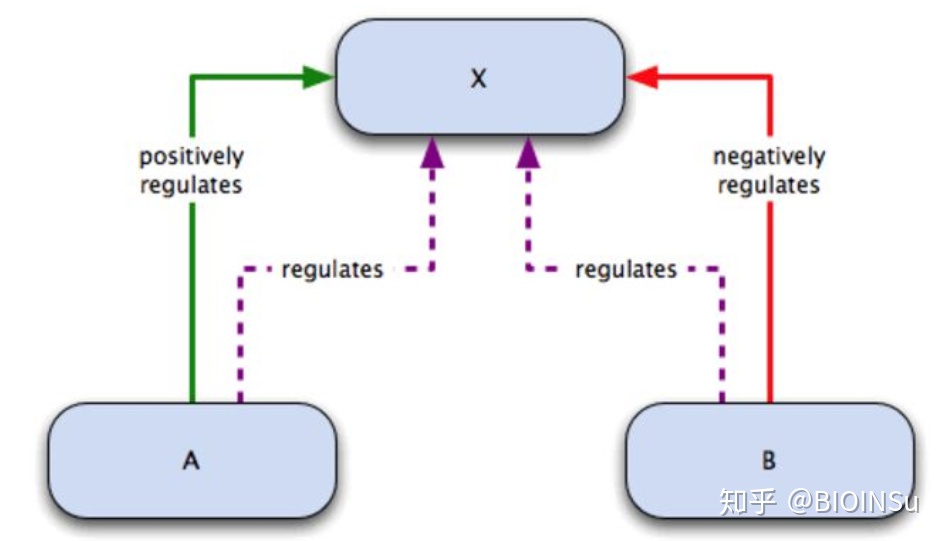
对于调控关系而言,有正调控和负调控两种。
GO Term
- GO Terms
GO Terms用于描述基因产物的功能。所有GO Terms的信息可以通过以下链接进行下载:
http://www. geneontology.org/page/d ownload-ontology
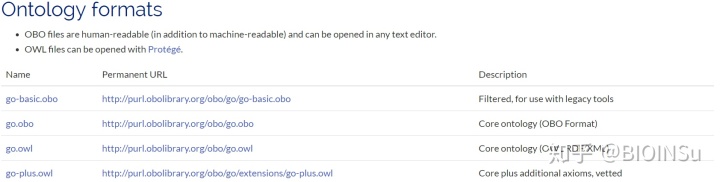
GO数据库的官网提供了obo, owl等格式 。
- GO Slim
GO本身是一个跨物种的概念,官方提供的GO文件涵盖了大部分的物种的所有功能。但是对于特定领域的物种来说,这些物种的基因的功能只占整个GO的一小部分,因此,需要从整个GO中抽取出一个简要的子集来省略掉一些详细的细节,这个子集就叫做GO Slim。对于特定的物种,采用合适的GO Slim, 可以有效减少后续数据挖掘的工作量。
在以下链接中,提供了不同领域的GO Slim:
http://www. geneontology.org/page/g o-subset-guide
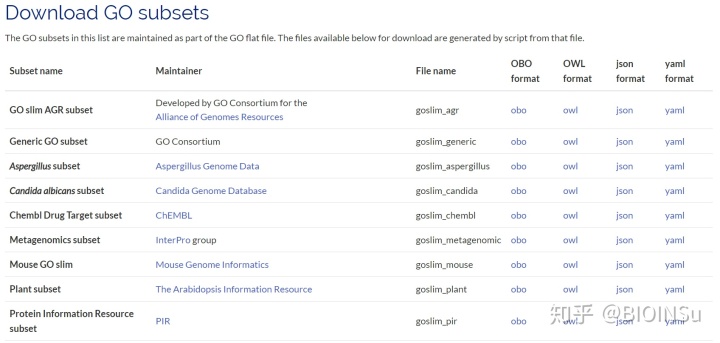
- 单独的GO Term
官网还将部分物种的GO注释信息单独抽出来提供下载,链接如下
http://www. geneontology.org/page/d ownload-go-annotations
- GO与其他数据库映射
除此之外,还提供了GO与其他数据库,比如KEGG Pathway, RDP, COG等数据库的映射关系,链接如下
http://www. geneontology.org/page/d ownload-mappings
例如,InterPro与GO的映射关系示例如下:
InterPro:IPR000003 Retinoid X receptor/HNF4 > GO:DNA binding ; GO:0003677
InterPro:IPR000003 Retinoid X receptor/HNF4 > GO:steroid hormone receptor activity ; GO:0003707
InterPro:IPR000003 Retinoid X receptor/HNF4 > GO:zinc ion binding ; GO:0008270
InterPro:IPR000003 Retinoid X receptor/HNF4 > GO:regulation of transcription, DNA-templated ; GO:0006355
InterPro:IPR000003 Retinoid X receptor/HNF4 > GO:nucleus ; GO:0005634基因富集分析方法
富集分析通常是分析一组基因在某个功能上是否过表达 (over presentation)。分析的结论是基于一组相关的基因,因此基因富集分析增加了研究的可靠性以及能够识别出与生物现象最相关的生物过程。
方法分类
基于数据来源和算法可将富集分析方法分为四大类:
- ORA法
Over-representation Analysis 过表达分析,又称为"2X2法"
具体步骤:
1.获得一组感兴趣的基因(一般是差异表达基因)。
2.给定的基因列表与某个通路中的基因集做交集,找出其中共同的基因并进行计数(统计值)。
3.利用统计检验的方式来评估观察的计数值是否显著高于随机,即待测功能集在基因列表中是否显著富集。
(最常用的统计检验包括:超几何分布、卡方检验、二项分布。)
优点:
基于完备的统计学理论, 结果稳健、可靠。
缺点:
(1)仅使用了基因数目信息,而没有利用基因表达水平或表达差异值,为了获得感兴趣或者差异表达基因,需要人为的设置阈值;
(2)ORA法通常仅使用最显著的基因,而忽略差异不显著的基因。在获得感兴趣的基因时, 往往需要选取合适的阈值, 有可能会丢失显著性较低但比较关键的基因, 导致检测灵敏性的降低;
(3)将基因同等对待,ORA法假设每个基因都是独立的,忽视了基因在通路内部生物学意义的不同(如调控和被调控基因的不同)及基因间复杂的相互作用;
(4)ORA假设通路与通路间是独立的,但这个前提假设是错误的。
- FCS法
Functional Class Scoring 功能集打分
具体步骤:
1.根据案例和对照状态下的基因表达谱对基因组中所有基因表达水平的差异值进行打分或排序,或直接输入排序好的基因表达谱。
2.把待测基因功能集中的每个基因的分数通过特定的统计模型转换为待测基因功能集的分数或统计值。
3.利用随机抽样获得的待测基因功能集统计值的背景分布来检验实际观测的统计值的显著水平,并判断待测基因功能集在案例和对照实验状态下是否发生了统计上的显著变化。
优点:
相较于ORA 法在理论上有明显突破, 考虑到了基因表达值的属性信息, 以待测基因功能集为对象来进行检验, 也使得检验结果更加灵敏。
缺点:
(1)仍独立分析每一条通路,但同一个基因可能涉及多条通路,所以不同通路间的基因出现重叠,所以别的通路可能由于重叠的基因,也出现显著富集;
(2)仍然把待测基因功能集中的每个基因作为独立的个体, 忽略了基因的生物学属性和基因间的复杂相互作用关系。
- PT法
Pathway Topology 通路拓扑结构
把基因在通路中的位置(上下游关系),与其他基因的连接度和调控作用类型等信息综合在一起来评估每个基因对通路的贡献并给予相应的权重,然后再把基因的权重整合入功能富集分析。不同的PT方法在具体的权重打分时,采用了不同的方式。
GO 等注释数据库中基因功能集中不包含任何拓扑结构信息,仅提供了可能属于同一通路的所有基因列表。所以,PT方法不能被用于GO通路的富集分析。
优点:
对于研究较完善、拓扑结构完整的通路,基于PT的基因功能富集算法会有更强的显著性。
缺点:
对于通路拓扑结构存在依赖性,对于研究较少、信息不完善的通路稳健性较差,目前通路注释的不完善。
- NT法
Network Topology 网络拓扑结构
目前NT法有一些不同的思路:
1.基于生物网络拓扑结构的富集分析方法,利用数据库中的基因相互作用关系来间接地把基因的生物学属性整合入功能的富集分析。
2.利用网络拓扑结构来计算基因对特定生物通路的重要性并给予相应的权重, 然后再利用传统的ORA 或 FCS 方法来评估特定生物通路的富集程度,如 GANPA 和 LEGO 等;
3.直接把基因列表中的功能富集问题利用网络转化为基因对的功能富集问题,如 NOA 等。
优点:
与传统方法相比,基于网络的基因功能富集分析方法加入了系统层面的基因重要性程度及关联信息,使得预测结果更加准确可靠。
缺点:
算法过于复杂,计算速度较慢。
富集分析的冗余性问题
目前几乎所有的功能富集方法都是对待测基因功能集进行独立检验, 而基因功能集之间存在较多的共同基因, 因而会导致富集的基因功能集之间出现冗余现象。
解决方法:
1.在富集分析时, 不把基因功能集进行独立检验, 而是把所有基因功能集作为一个整体来进行富集分析。
2.对获得的富集基因功能集进行聚类和过滤。先把基因功能集按照互相之间共同基因的重叠程度构建一个网络, 利用网络模块划分的方法得到一系列基因功能集模块, 使得每个模块内部的功能集具有较高的相似度。
GO富集分析
基本概念
利用GO数据库中的基因注释信息进行基因富集分析。GO富集分析的结果包括包括GO功能分类结果和GO功能富集结果。
GO功能分类:在某一功能层次上统计蛋白或者基因的数目或组成。
GO功能富集:获得相对于参照基因显著富集的功能类别。
分析原理
根据挑选出的差异基因,计算这些差异基因同GO分类中某(几)个特定的分支的超几何分布关系,GO 分析会对每个有差异基因存在的GO Term返回一个假定值p-value,小的p 值表示差异基因在该GO 中出现了富集。
GO富集分析的结果
GO富集分析结果主要由有向无环图DAG和柱状图两种形式所表示。
- DAG
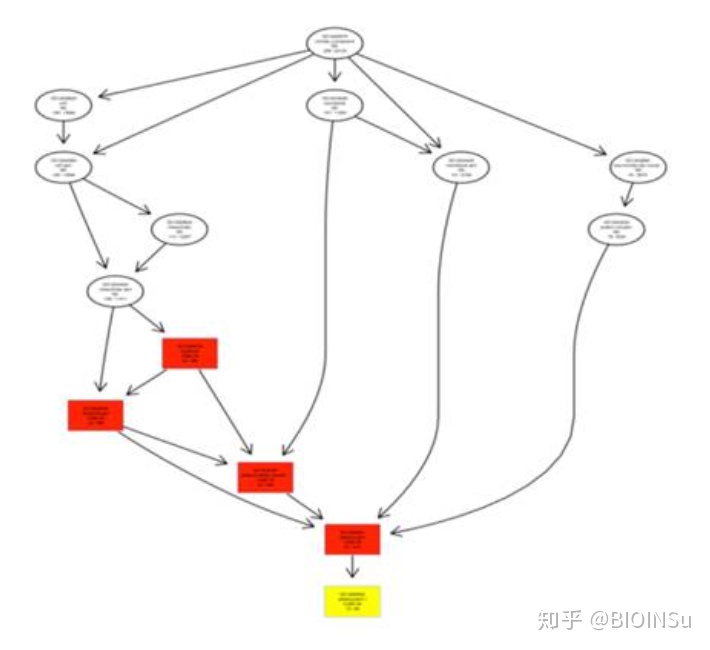


图形的颜色反应了基因在某一个GO Term上的富集程度,颜色越深代表富集程度越显著。GO Term的层级越低,功能描述越具体。
- 柱状图
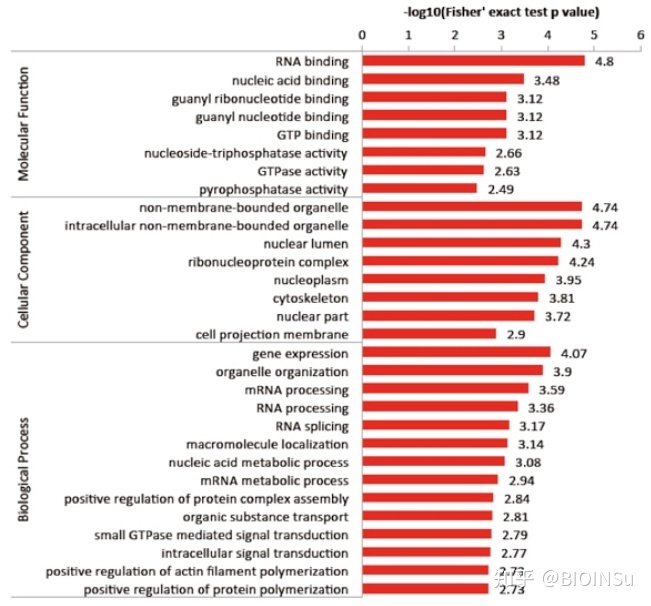
GO在线富集分析工具
有许多网站提供GO富集的功能,这些网站大多要求提交一组基因,然后通过在线查询的方式给出结果。
例如DAVID(The Database for Annotation, Visualization and Integrated Discovery ):
https:// david.ncifcrf.gov/
GO本地富集分析工具(GO::TermFinder)
使用本地的富集分析工具,能够减少大批量查询的时间。这里以GO::TermFinder为例实现本地GO富集分析。
1.安装GO::TermFinder :
操作系统:Ubuntu (Windows下的用户可以在Microsoft Store 下搜索Ubuntu,安装该应用程序后即可获得虚拟的Ubuntu系统进行操作)
GO::TermFinder:是Perl下的一个模块,可以对一组Genes和GO Terms做基因富集分析。
(1)首先在Ubuntu系统下安装好Perl解释器以及Perl的包管理器CPAN。安装完毕后确认CPAN能够使用: sudo perl -MCPAN -eshell 。打开成功后即为成功安装CPAN,键入q退出CPAN。
(2)在安装GO::TermFinder之前需要安装一些Perl下的依赖库,否则安装时会一直报错,具体如下:
- Storable:使用CPAN下install Storable即可安装
- CGI:使用CPAN下install CGI 即可安装。
- GD:首先需要在Ubuntu系统下安装zlib、libpng、libgd模块(可能可以仅安装个别,未验证但全部安装肯定可以),使用sudo atp-get update 以及 sudo apt-get install libgd2-xpm-dev 等命令 进行安装,然后再进入CPAN下install GD进行安装才能成功,否则直接CPAN下安装会报错。
- GraphViz:首先需要在Ubuntu系统下sudo apt-get install graphviz,然后再进入CPAN下install GraphViz进行安装才能成功,否则直接CPAN下安装会报错。
(3)安装完上述模块后入CPAN下install GO::TermFinder进行安装,如果还出现报错,则需要按照出错提示 安装对应的依赖模块才能够成功。
2.下载对应的ontology files:
GO::TermFinder主要需要两个与Gene Ontology有关的文件:
(1)obo文件,是用来存储所有GO term关系的文件,GOC上分有basic版本和不稳定版本,不稳定版本会经常拓展,但是包括了GO的其他关系。
(2)annotations文件(.gaf结尾),是用来存储gene 的GO 注释的文件 ,不同的物种有不同的annotation文件,在GOC上找对应的物种文件进行下载。
这两个文件均可以从Gene Ontology Consortium的官网上进行下载:
3.获取分析代码并进行分析:
可以在CPAN官网上下载GO::TermFinder的模块包:
里面examples文件夹中有一个其他团队写好的分析代码 batchGOView.pl 及其配置文件GoView.conf。该perl代码是利用GO::TermFinder模块对基因文件进行批量处理分析的代码,配置文件内能够对生成的图以及p-value阈值等进行设定。
分析步骤为:
(1)打开GoView.conf,更改:
- annotationFile:把对应值改成下载下来的GO annotations文件的位置。
- ontologyFile:把对应值改成下载下来的obo文件的位置。
- aspect:表示GO分析的类别。GO数据库被分成了三个大类,分别cellular_component, biological_process 和molecular_function,所以这里的参数有三种可能:C, P 或者 F,每次只能设置一个。
- pvalueCutOff:可以设置p-value的阈值。
(2)更改配置文件完成后,准备gene文件(因为是批量处理,所以可以是多个),gene以txt文本的形式存储, 每行存储一个gene,放在对应的位置。
(3)此时再次确保 batchGOView.pl、GoView.conf、obo文件(.obo)、annotations文件(.gaf)、gene.txt文件都已经存在。
(4)在命令行下运行命令 perl batchGOView.pl GoView.conf genes.txt genes2.txt ...... 进行富集分析。
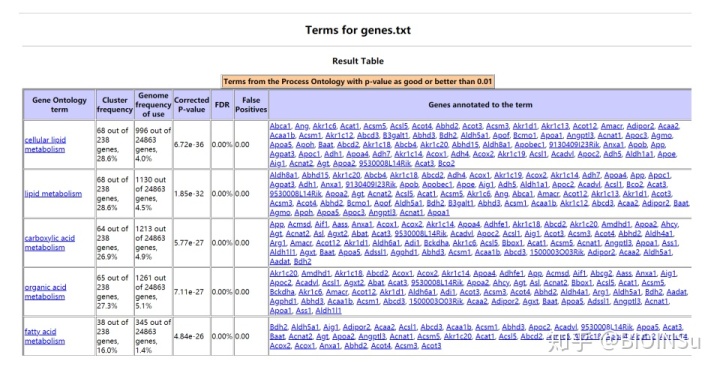
(5)成功运行后,每个gene.txt文件都会对应生成一个html文件,里面即为富集分析的结果。
结果示例:
参考资料
富集分析:
https://max.book118.com/html/2018/0423/162698642.shtm
http://www.360doc.com/content/17/0919/01/47411701_688261255.shtml
http://ibi.hzau.edu.cn/sysbio/stuPDF/2010-3.pdf
GO数据库:
https://www.jianshu.com/p/b72363ac4840