一个菜鸟的进阶之路
一个菜鸟的进阶之路
写下这篇文档! 记录学习生信学习,包括Linux、python、R语言、perl、qiime2等软件的经历和问题吗,也包含了一些基本作图方法和生物系统进化,以及微生物学的知识。
关注微信公众号 国科大青年

第一章 Linux
Linux服务器
由于所研究的课题需要进行数据分析,所以开始捣鼓服务器。
1.Window下xmanager的安装
2.用Xshell连接服务器
3.服务器基本命令
3.1vim 命令
常用vim命令
vim命令和操作合集:
原文链接:https://blog.csdn.net/weixin_37657720/article/details/80645991.
3.2top命令
常用的top命令
top命令和操作集合:
原文链接:https://blog.csdn.net/yjclsx/article/details/81508455.
3.2文件查找命令
4.如何管理好你的服务器
4.1.软件的安装管理
4.2.数据管理
linux系统
1.认识linux系统和linux入门
2.linux软件管理
3.shell语言
4.linux下shell命令
5.linux
虚拟机
1.bio-linux
2.UOS
3.depin
第二章 python
Window下python的安装
python学习书籍和课程
一些简单脚本的撰写
用python进行数据分析
用python画图
第三章 R语言
从需求出发学习R语言
当你需要做一个图时,或者发现一个好看的图需要模仿时,
用R语言就可以了!
用R语言就可以了!
用R语言就可以了!
这句话总是在耳边回荡。
你百度了一下,似乎只需要复制一下代码运行一下就可以了。
但是,仔细一想又无从下手。
这时的你是如何的孤独和无助
你的需求
1.文章中的多种图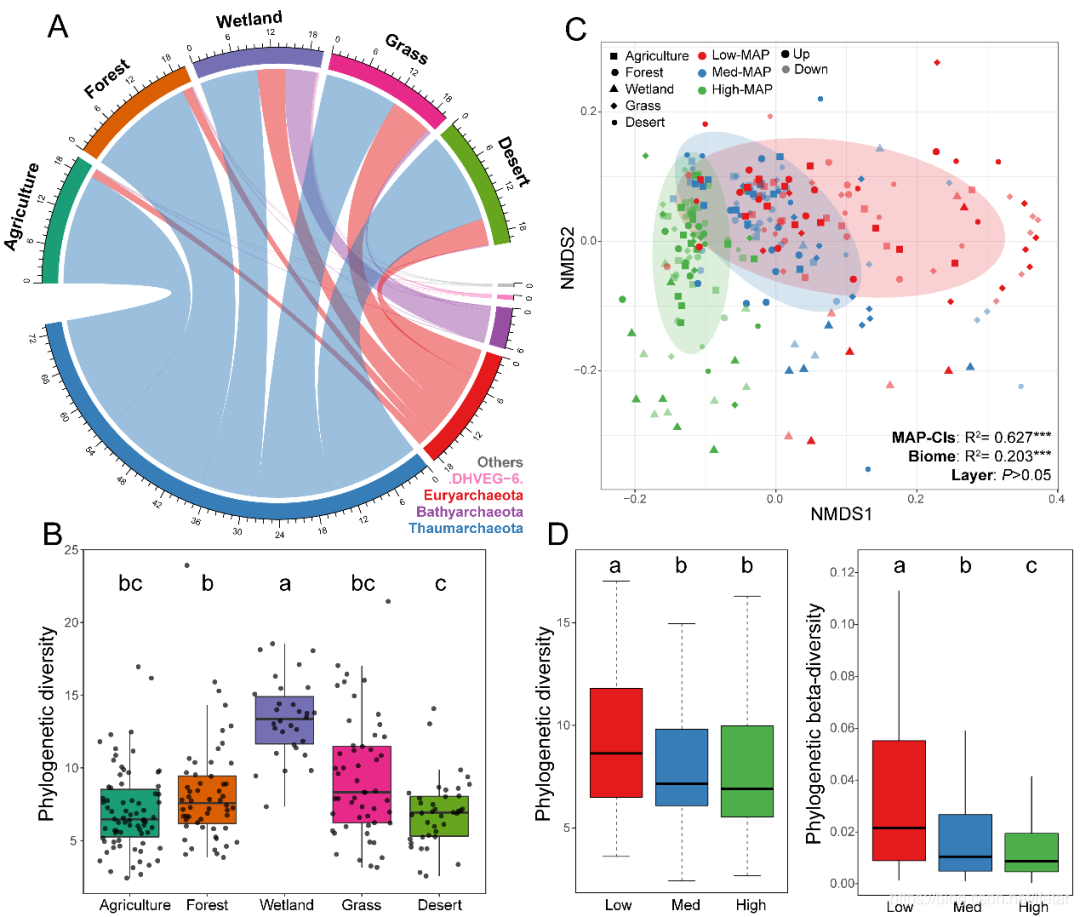
2.环形图
为了绘制出图二的环形图,你百度了一下,可能找到了如下代码:
# library
library(circlize)
# Arrange the layout
layout(matrix(1:9, 3, 3))
# A loop to create 9 circular plots
for(i in 1:9) {
par(mar = c(0.5, 0.5, 0.5, 0.5), bg=rgb(1,1,1,0.1) )
factors = 1:8
circos.par(cell.padding = c(0, 0, 0, 0))
circos.initialize(factors, xlim = c(0, 1))
circos.trackPlotRegion(ylim = c(0, 1), track.height = 0.05, bg.col = rand_color(8), bg.border = NA )
# add links
for(i in 1:20) {
se = sample(1:8, 2)
circos.link(se[1], runif(2), se[2], runif(2), col = rand_color(1, transparency = 0.4))
}
circos.clear()
}
但问题是你需要怎么绘制这个图尼?
复制代码,然后运行?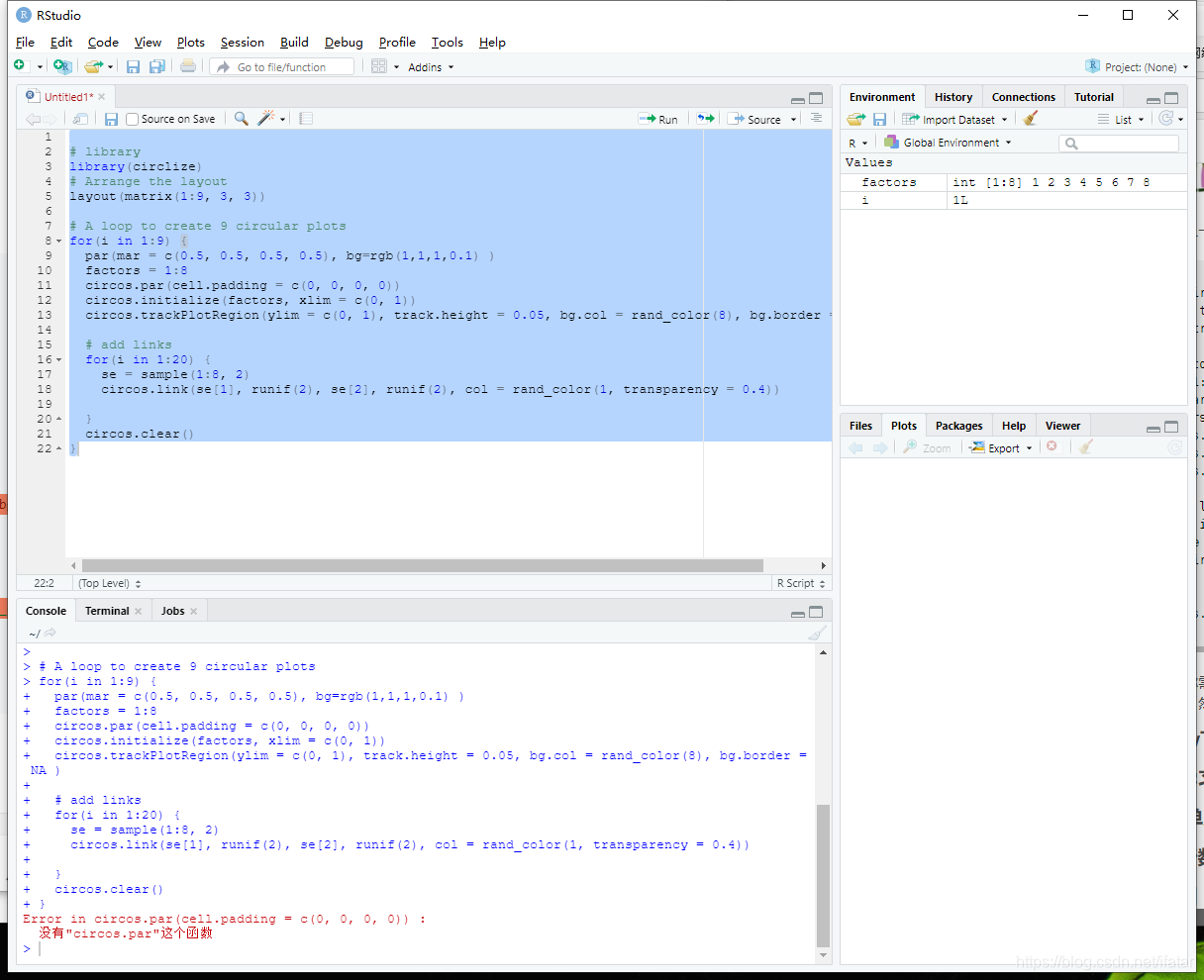
你复制了也运行了,发现什么都没有。
没有安装R和Rstudio请直接查看相关的安装教程
为什么尼?
为了回答这个问题,我们需要回归到代码中寻找答案。
# library
# 井号这个符号表示注释,不参与运行
library(circlize) #告诉我们需要加载一个包,如果没有这个包就得先安装之后再加载
# install (" circlize")
# library (circlize)
# Arrange the layout 根据我们的需要准备一个布局,如图所示我们需要构建一个3X3 的图形矩阵
layout(matrix(1:9, 3, 3))
# A loop to create 9 circular plots 进行一个循环创建9个环形图,
for(i in 1:9) {
par(mar = c(0.5, 0.5, 0.5, 0.5), bg=rgb(1,1,1,0.1) )
factors = 1:8
circos.par(cell.padding = c(0, 0, 0, 0))
circos.initialize(factors, xlim = c(0, 1))
circos.trackPlotRegion(ylim = c(0, 1), track.height = 0.05, bg.col = rand_color(8), bg.border = NA )
# add links 给绘制的图形添加线,
for(i in 1:20) {
se = sample(1:8, 2)
circos.link(se[1], runif(2), se[2], runif(2), col = rand_color(1, transparency = 0.4))
}
circos.clear()
}
#这样我们的图形就绘制完成了。
但是这样就存在了一个问题,如何用自己的数据怎么绘制图形尼?
因此你首先要知道什么是数据以及这个图需要什么样的数据!
为此我们给出了以下的图,帮助你更好的理解什么是数据。
library(circlize)
# 模拟数据集,创建一个数据集采用的是赋值法,换句话说就是数据等于什么,其由那几部分组成。列如以下数据由三部分组成,factor和x以及y三部分。
data = data.frame(
factor = sample(letters[1:8], 1000, replace = TRUE),
x = rnorm(1000),
y = runif(1000)
)
# factor由
circos.initialize( factors=data$factor, x=data$x )
# 创建区域.
circos.trackPlotRegion(factors = data$factor, y = data$y, panel.fun = function(x, y) {
circos.axis()
})
# 添加散点
circos.trackPoints(data$factor, data$x, data$y, col = "blue", pch = 16, cex = 0.5)
Window下R的安装
R语言学习书籍和课程
一些简单脚本的撰写
用R进行数据分析
用R画图
第四章 Perl
Perl环境的搭建
Perl语言学习书籍和课程
一些简单脚本的撰写
用Perl进行数据分析
用Perl画图
第五章 qiime1.9.1和qiime2
从双端数据开始分析(pair fastq原始数据)
两端序列从头分析 https://blog.csdn.net/leadingsci/article/details/80772454
从fasta文件文件开始分析
qiime1.9.1的安装和环境
1.软件安装
#qiime1.9.1
conda install qiime
#flash1.2.1
conda install flash
#usearch
熟悉qiime1.9.1的分析流程——数据准备
- 数据和文件准备
1.fasta格式原始数据
2.map文件
用示例数据进行分析
用自己的数据进行分析
qiime1可视化
qiime2的安装和环境
认识qiime2和熟悉qiime2的分析流程
用示例数据进行分析
用自己的数据进行分析
qiime2可视化
第六章 16S数据分析
16S的基本概念
16S的技术进展
16S的数据分析流程
查询某一种细菌的拷贝数
https://rrndb.umms.med.umich.edu/
在这个网址中输入具体的细菌名字就会有相应的细菌的拷贝数,可能是属水平,但是种水平相对较少。
用自己的数据进行16S分析
16S相关软件
1.picrust2功能预测
1.1认识PICRUST2
项目简介https://github.com/picrust/picrust2/wiki
PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States)是一款基于标记基因序列来预测功能丰度的软件。
1.2安装PICRUST2
1.3分析流程
https://www.jianshu.com/p/e3beaee77423.
一步完成分析
文章链接: https://blog.csdn.net/weixin_46880672/article/details/105583296.
分步骤完成分析
文章链接: https://github.com/picrust/picrust2/wiki/PICRUSt2-Tutorial-(v2.1.4-beta).
第七章 宏基因组数据分析
宏基因组的基本概念
宏基因组的技术进展
宏基因组的数据分析流程
用自己的数据进行宏基因组分析
宏基因组相关软件
第八章 全基因组数据分析
从零开始完整学习全基因组测序(WGS)
1.从他人的博客中认识全基因组数据分析流程
第1节 测序技术
文章链接:https://www.plob.org/article/11652.html?wpzmaction=add&postid=11652.
第2节 FASTA和FASTQ
文章链接:https://www.jianshu.com/p/50ff302d049f.
第3节 数据质控q
文章链接:https://www.jianshu.com/p/36891a89ed6e?utm_campaign=hugo.
第4节 构建WGS主流程
文章链接:https://www.plob.org/article/11698.html.
2.全基因的基本概念
细菌基因组的研究方法总结
https://www.sohu.com/a/344522967_278730
1.细菌基因组分析的研究价值
1)发现新物种(新功能)
在微生物群落研究异常火爆的今天,仍然有里程碑式的成果离不开单菌基因组的研究。比如,硝化过程的两步反应(氨氧化反应、亚硝酸盐氧化反应),100多年来都被认为是由两类微生物分别完成(氨氧化菌和亚硝酸盐氧化菌),直到近几年因一种细菌的基因组被组装出来,才发现它的基因组上两种反应的酶基因都有。两步反应由同一种微生物催化的成果(如图1)推翻了以往认知的同时,也表明了单菌组装的重要价值。另一方面,针对新物种(数据库中没有其基因组信息)的基因组序列,可以开展更丰富深入的研究。

硝化微生物特征
2)基于基因组组分的功能研究
获得目标细菌的基因组序列,就相当于有一把打开该物种神秘世界的钥匙。基于序列信息,结合数据库和预测软件,即可对其基因组进行破译,涉及基础功能代谢、致病性、耐药性、次级代谢产物资源……等多个方面。
3)基于组分间位置关系的功能研究
我们不仅可以从独立的基因层面了解物种的功能,还可以结合基因间的位置关系分析推测基因的功能特征、基因迁移规律等。比如根据基因组上目标基因上下游的特征,推测致病性、抗药性迁移规律(如图2)。
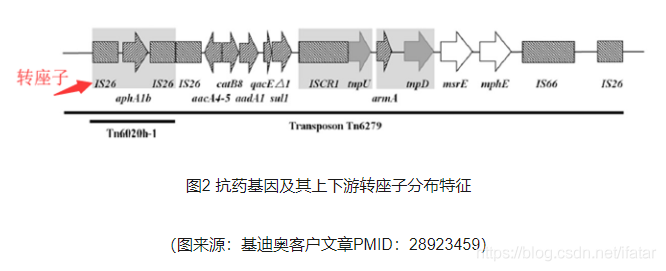
抗药基因及其上下游转座子分布特征
(图来源:基迪奥客户文章PMID:28923459)
4)基于基因组间序列比较的研究
获得目标菌株基因组后,可以从数据库下载近缘物种基因组序列,开展物种间进化关系研究,比如系统发育树;物种间功能差异、基因结构差异研究,比如基因组共线性分析,基因家族分析等。
2
细菌基因组组装流程的发展完善之路
三代测序的出现,带领细菌基因组突破了二代序列组装拼接不完整的瓶颈。三代Pacbio超长reads可以轻松实现细菌基因组“0 gap”的组装。后来,由于三代测序准确率相对较低,为了获得更科学可靠的基因组序列,又发展出了三代与二代相结合的方式,主要有两种类型,1)二代reads对三代长reads校正,然后使用校正的三代reads组装;2)三代reads组装后,使用二代reads对基因组校正。至此,细菌基因组研究已经发展出了较为完善可靠的体系。
针对细菌三代测序项目,基迪奥免费赠送二代Illumina测序数据和校正分析服务,在三代组装完成后补充二代reads比对校正,让数据挖掘更可靠!
3
细菌基因组主要分析方法
通过数据库注释和软件预测这两种方式,对组装获得的细菌基因组序列进行破译,以适用于更广泛的物种生态学功能、致病性等研究。
3.1 基因组组分预测
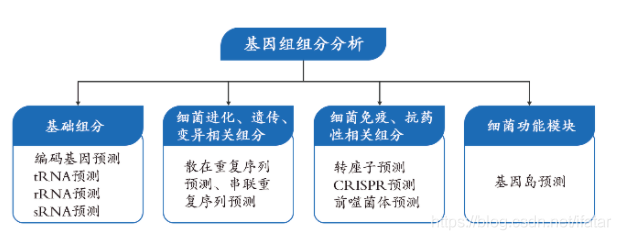
组分预测总结
3.2 数据库注释
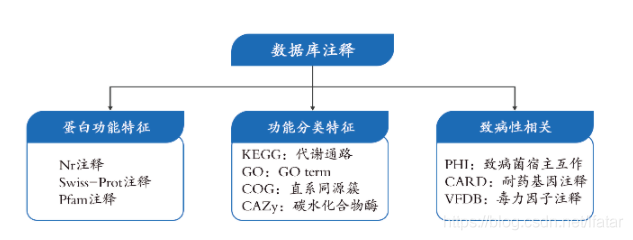
数据库注释总结
3.3 特色功能分析
1)致病性相关:分泌蛋白预测、TNSS效应蛋白预测
2)蛋白结构分析:信号肽预测、跨膜蛋白预测
3)群体感应、耐药性、环境压力响应等相关:双组分系统预测(图5)
4)次级代谢产物:次级代谢产物基因簇预测
3.基因组共线性
1.使用Mauve分析基因组共线性
参考文档https://mp.weixin.qq.com/s/mzrxGhGCr7Xj-g7O1az8zg
参考视频https://www.bilibili.com/video/av584030222/
下载安装
http://www.darlinglab.org/mauve/mauve.html
安装之前需要安装java,尽量安装相对较低的版本,因软件日期较久远,新版本java可能不匹配,出现安装错误。
java安装网址
https://www.oracle.com/java/technologies/javase-downloads.html
给的建议是先安装最新版本的java,然后配制环境变量,最后下载之前版本的java并安装,然后安装mauve软件。
文件准备
下载相应文件,本文采用细菌基因组文件用于执行后续分析和演示
自己的基因组
下载的基因组
如何下载基因组文件
在window系统下操作
首先,点击菜单栏“File”>“Align sequence…”或者“Align with progressiveMauve…”,在新的窗口中点击“Add Sequence…”选择将基因组序列文件读取。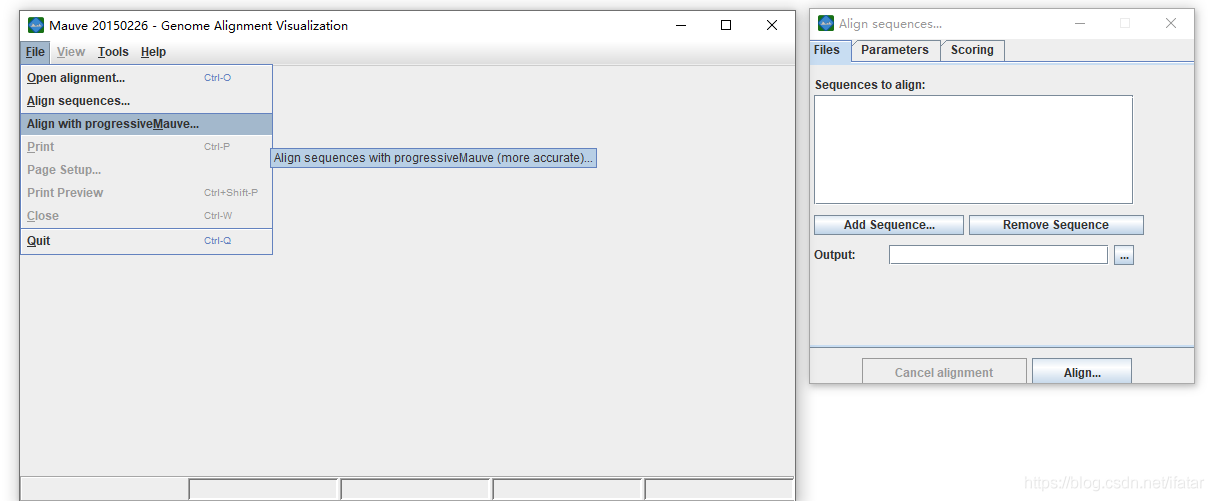
然后将文件拖入右侧白框内
随后,在窗口的“Parameters”或“Scoring”界面中,可以对基因组比对等程序运行参数进行调整。
参数详情可参考官方文档:http://www.darlinglab.org/mauve/user-guide/aligning.html
设置完毕后,在窗口的“Files”界面的“Output”选项中指定结果的输出路径,最后点击“Align…”运行共线性分析。等待一会儿后,最后获得的共线性分析结果界面如图所示。
然而
这是由于你的数据格式不正确导致的
输入解压后的正确格式数据后得到如下结果
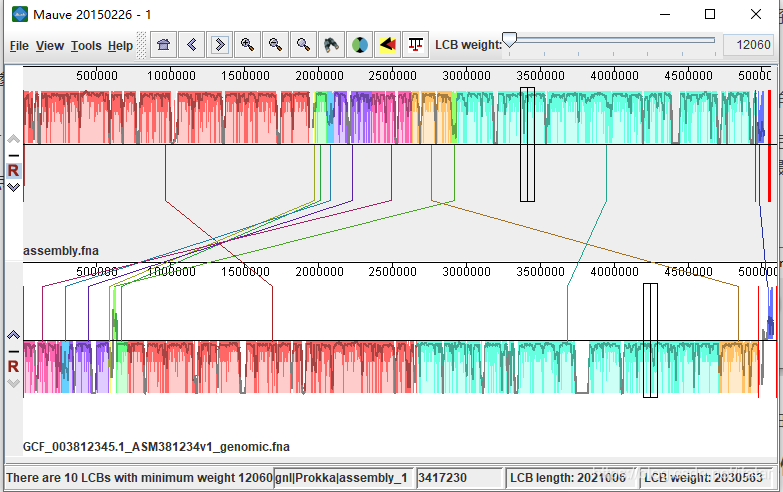
通过菜单栏“View”,可以对共线性图谱进行可视化细节部分的调整。
如果期望将图片输出,可点击菜单栏“Tools”>“Export”>“Export Image…”即可将当前界面输出为jpg、png等位图格式。若想获取矢量图,可点击菜单栏“File”>“Print…”输出为pdf格式。
很多文献中,还选择将Mauve输出的共线性图谱和其它分析作个结合,如和物种进化树放在一起,更形象地为读者呈现出基因组相似程度以及进化动力学信息。
此外,“Tools”>“Export”还提供了其它输出项,可用于查看基因组之间的结构差异以及进化事件等。
用示例数据进行分析
认识结果数据和对数据结果进行解释
用自己的数据进行分析
1.数据的准备
2.数据的分析
进一步优化结果
第九章 作图软件
TBtools
CorelDRAW
inkscape
AI
1.数据的准备
2.数据的分析
PS
cytoscape
1.数据的准备
2.数据的分析
PS
第十章 生物系统进化
各种生物系统进化树的认识和制作
图一
图二
图三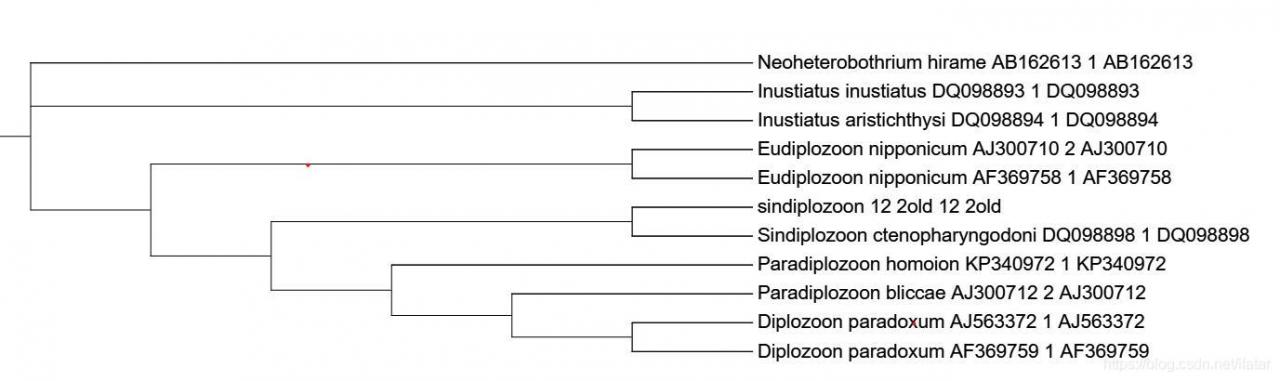
图四
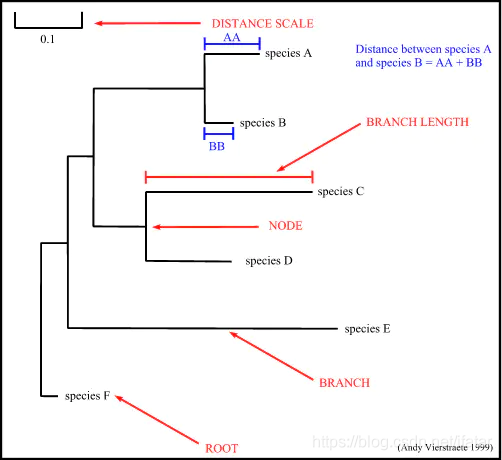
这里分享一篇别人总结得比较好的文章
文章链接:http://www.360doc.com/content/17/1226/18/33459258_716501139.shtml
一个系统进化树中包含着大量的有关不同物种之间进化关系的信息,而解码这些信息并不容易,多数情况下需要理解进化树中包含哪些单元以及他们各自代表什么含义。
1、进化树单元
图1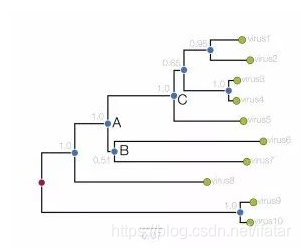
如上图所示就是一个经典的树杈状系统进化树
2、进化树中包含的信息
下面跟着小编一一认识一下每个单元的含义(以上图为例)
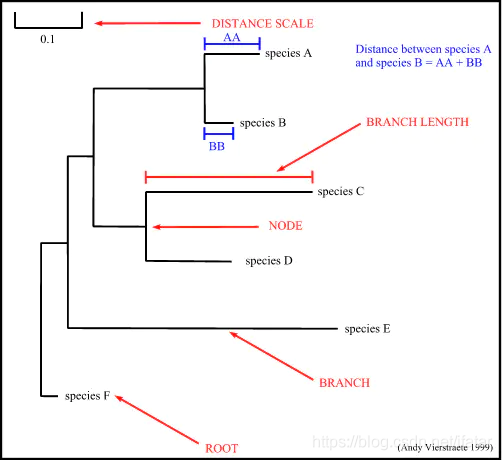
(1)水平方向:遗传变量的变化程度
水平方向上的分支代表进化谱系随着时间的变化,分支长度越长表示该分支对应物种的变化越大。
(2)标尺:底部的短线
标尺下部的数字0.07指该长度的分支代表基因组的遗传变异度为0.07。其中,遗传变异度通常表示基因组中每个位点碱基的替换频率,计算公式为:变异度=变异碱基数/总碱基数(%)。通常情况下会以百分比的方式展示,进而可解释为每100个碱基位点的变异度。
(3)垂直方向:无特别含义
其长度无关紧要,通常是为了调节不同分支间的大小以免不同标签之间的重叠,提升树图的美感及可读性。当然,我们还可以从垂直方向看出来不同物种之间进化关系的远近,在树枝上挨得越近,就说明这两个基因组,也就是他们所代表的物种进化分歧时间更短,亲缘关系更近。
(4)节点:彩色圆圈
进化树上的节点有外部节点和内部节点两种。外部节点又称为叶节点(上图中绿色圆圈),代表参与分析的基因组数据(例图中是病毒的基因组序列),一般情况下一个基因组也即带代表一个物种。内节点用蓝色的圆圈表示假定祖先。
进化树的根(红色圆圈)告诉我们终极祖先的位置,基于这个点依据不同分支在水平方向上的长度可以对不同物种近似进化时间进行排序。例如:在上图中我们可以说A物种比B和C物种在进化时间上更早,同时也可以认为水平方向上进化时间从左到右依次增加。
(5)分支
因为分支长度代表基因序列相似度,只能代表近似进化时间。因此,需要根据基因序列相似度与进化时间假说对这种进化距离进行转换,即分子钟。
(6)Bootstrap值
图中每个节点左边的灰色数字代表该节点的bootstrap值。该值范围从0-1,也可用百分比的形式代表,值越大该节点置信度越高。
(7)拓扑结构
图2
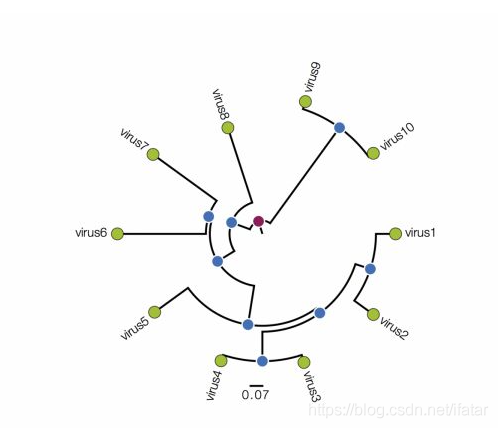
图3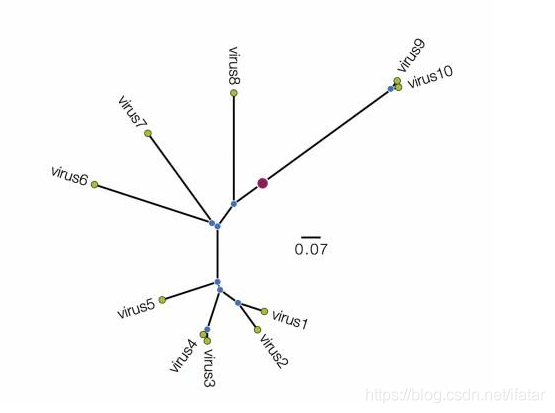
除了图1中的经典展示形式外,系统进化树也可用不同的拓扑结构进行展示,如图2所示的圈图,以极坐标的形式展示。垂直方向代表角度,水平方向代表叶节点与中心节点的距离。这种展示形式从视觉上来讲比较震撼,尤其在物种数比较多的情况下,更显优势,但实际可读性不佳,尤其是对于进化距离的评估。图3是一个辐射树,经常用于根节点不确定的进化树构建,这种树可以有效地将相似度高的序列紧密聚集在一起,因此容易将这些序列之间的细致信息模糊化。
基于不同树型展示结果的差异,图2这种拓扑结构的进化树多用于差异较大的物种,而图3更适合做亲缘关系较近物种的进化树
今天的主要内容就介绍到这里,不知道对不对你的口味,欢迎在评论区与小编互动哦!
了解了以上的基本内容后,那么尝试理解一下以下内容:
最后的最后,再给大家明确系统进化分析中容易混淆的几个概念
(1)Dendrogram、cladogram和phylogram的区别(仅在部分领域适用)
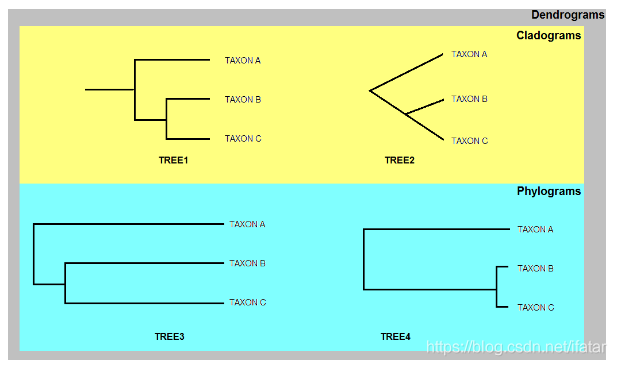
Dendrogram:包括cladogram和phylogram
cladogram:分支仅代表进化关系,不代表进化时间,TREE1 = TREE2
phylogram:分支代表进化关系和时间, TREE3 ≠ TREE4
(2)有根树和无根树:
有根树(TREE1):能够从树中找到物种的共同祖先,以二进制树展示
无根树(TREE2):不能从树中找到物种的共同祖先,树中一个节点有三个分支,其余节点为两个分支。
(3)梯状树和枝状树:
相对与梯状树(TREE1 &TREE3),远源关系的物种更适合用枝状树(TREE2&TREE4)构建进化树。