写在前面的话
前面我们说到,HOMER软件可以进行多种类型的motif分析,如 promoter motif analysis ,基因组位置motif分析(ChIP-seq分析中的motif分析),利用自定义的fasta文件进行motif分析,RNA序列的motif分析(分析CLIP-seq数据中的RNA binding elements)。 但是,不管我们如何调用HOMER,对于发现motif的基本步骤都是一致的,下面主要介绍一下各步骤的原理:
原理理解即可,HOMER已经将各个步骤封装起来,不用一步一步分别进行。
预处理
1. 序列提取
若给出的基因组位置信息,则提取出来的是对应的基因组序列;
若给出的是基因accession number,则需要选择适当的promoter区域
2. 背景选择
如果没有自定义背景序列信息(即:-bg 参数),HOMER将自动为用户选取。如果使用的是基因组位置,将从整个基因组序列抓取GC含量一致的序列作为背景序列。若进行的是promoter分析,则将所有的promoter作为背景。
3. GC含量矫正
将目标序列和背景序列对GC含量进行分bin(5%区间),背景序列通过调节权重得到和目标序列同样的GC含量分布。这可以使得HOMER在分析来自CpG岛时的序列时,不会简单的找到那些GC富集的motif。下图是一个ChIP-seq分析中GC含量分bin的例子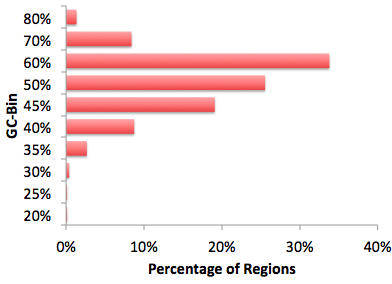
4. 自动标准化
除了GC含量之外,目标序列还会存在其他的情况影响分析结果。如外显子上的密码子偏好等生物学现象,由A富集序列优先排列引起的实验偏差。当这些偏差显著时,将会影响HOMER的motif分析。HOMER通过给背景序列分配适当的权重,减少寡核苷酸序列频率(如AA)在背景与目标的差异。
重头预测motif
默认情况下HOMER调用homer2版本,若想使用旧版本,在命令行中添加
-homer1参数即可
5. 解析输入序列
根据设置的motif长度,将输入序列解析为寡核苷酸,并生成一个寡核苷酸频度表。该表只记录出现的寡核苷酸和其出现的次数,可以增加motif搜索时的效率,但是也会破坏寡核苷酸与其原始序列的关系。
6. 寡核苷酸自动均一化
HOMER除了在第四步中对全局序列进行了自动均一化矫正,还在寡核苷酸频度表中进行了矫正。该方法的主要思想是将那些较短的寡核苷酸与较长的寡核苷酸相平衡,但是该方法由于存在许多与motif同长的寡核苷酸,需要分配数量众多的权重。对于那些极端的序列偏好,该方法不会移除它们。
7. 全局搜索
在构建好寡核苷酸频度表后,HOMER进行全局motif搜索。其基本原理就是若某个motif富集,则其存在的寡核苷酸也同样富集。为了增加灵敏度,HOMER允许搜索motif时的错配,同时跳过多个错配的寡核苷酸节省计算资源。
8. 矩阵优化
9. Mask and Repeat
当最优oligo被优化成motif后,motif 对应的序列从要分析的数据中移除,接下来再分析最优的…..直到设定个数的motif被发现。
搜索已知的motif(homer2版本)
10. 加载moitf数据库
HOMER从以前的数据中加载一个已知的motif列表从而搜索已知的motif还可以通过在命令行中指定的motif( -mknown)或通过编辑homer包中的文件( data/knownTFs/known.motifs )来添加motifs。
11. 扫描所有的motif
HOMER通过扫描所有的序列,确定每个motif的富集度,并计算其显著性。
motif输出的结果分析
12. Motif文件
HOMER的输出有许多后缀名为 motif 的文件,其内容如下: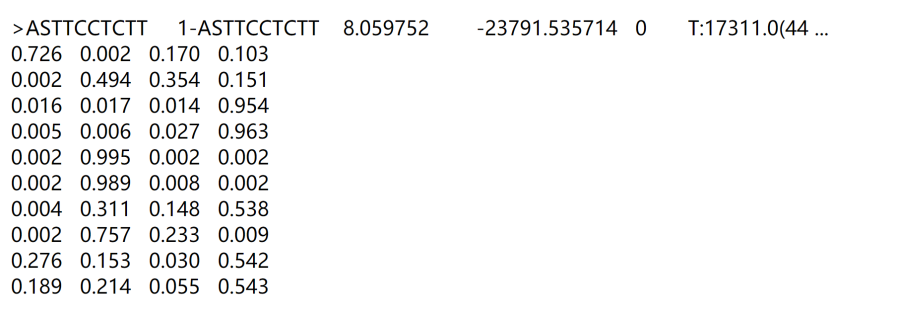
每个motif信息是一块,均以>开头。>所在行的信息以tab分隔。motif首行信息解释:
>+ 一致性序列:如图上的>ASTTCCTCTTMotif名称:如图上的1-ASTTCCTCTT
比值检测概率的log值:8.059752
P-value得log值:-23791.535714
占位符:如上图的0,不具有任何信息
逗号分隔的富集信息,如:T:17311.0(44.36%),B:2181.5(5.80%),P:1e-10317
T表示带有该motif的目标序列在总的目标序列(target)中的百分比B表示带有该motif的背景序列在总的背景序列(background)中的百分比P表示最终富集的p-value
逗号分隔的motif统计信息,如:Tpos:100.7,Tstd:32.6,Bpos:100.1,Bstd:64.6,StrandBias:0.0,Multiplicity:1.13
Tpos:motif在目标序列中的平均位置
Tstd:motif在目标序列中位置的标准差
Bpos:motif在背景序列中的平均位置
Bstd:motif在背景序列中位置的标准差
StrandBias: 链偏好性,在正义链上的motif数与反义链motif数的比值的log
Multiplicity:具有一个或多个结合位点的序列中每个序列的平均出现次数
13. denovo预测的motif
首先对产生的motif进行去冗余,然后将motif转换为向量值从而计算皮尔逊相关系数。HOMER产生的motif结果使用网页展示,该文件名称为 homerResults.html ,而目录homerResults/ 存放着所有该网页依赖的文件和图片。生成的motif首先去冗余。以下是一个简单的输出示例: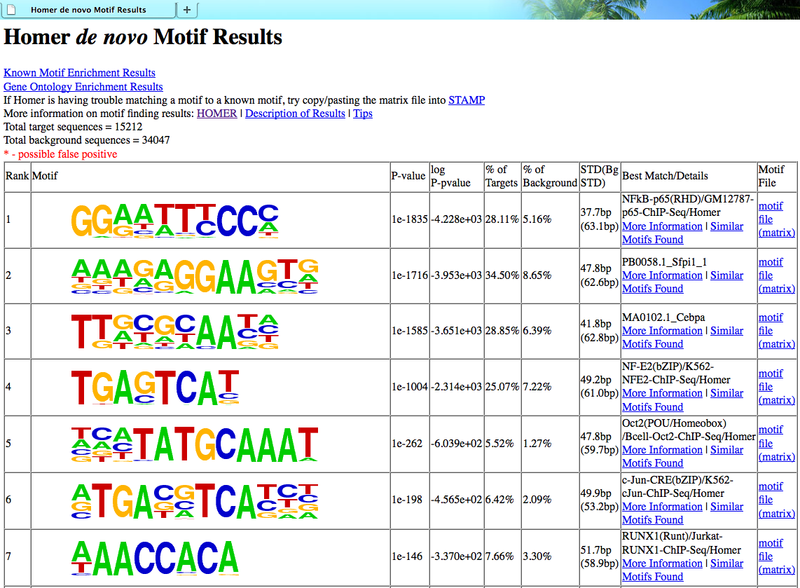
输出结果按照p-value排序,最后一列是一个链接到motif文件的超链接,可以从这个文件中找到包含此motif的其他序列。在Best Match/Details 列中,HOMER将会展示与denovo motif最匹配的已知motif(不可全信,最匹配不一定是最优)。该列还包含有一个名为 More information 的超链接,点击后会出现以下页面:
该页面包含motif的一些基本信息,如链接到motfi文件的超链接,且可以生成motif logo的pdf格式。
接着是与已知motif的match。这部分主要看看denovo motif和已知的motif的相似性,需要检查一下是否合理,如下: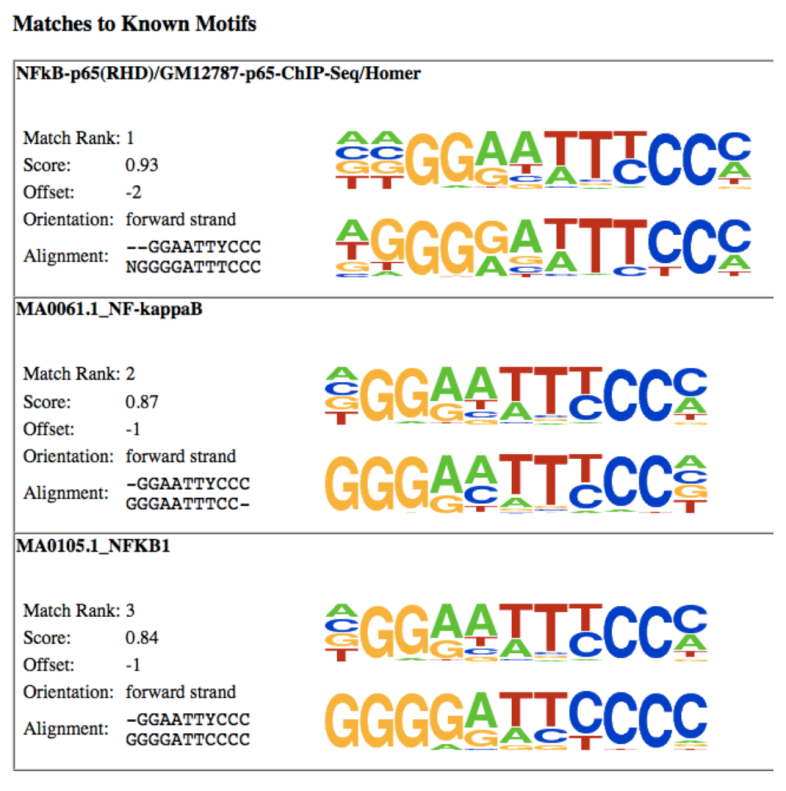
如果点击 Simiar motifs 超链接,将会展示在denovo motif搜寻过程中与该motif相似的其他motifs(这些motif的enrichment value相对较低)。通常我们会点击查看一下是否有一些motifs被不正确的分类,比如几个motif具有几个相同的碱基。具体界面如下:
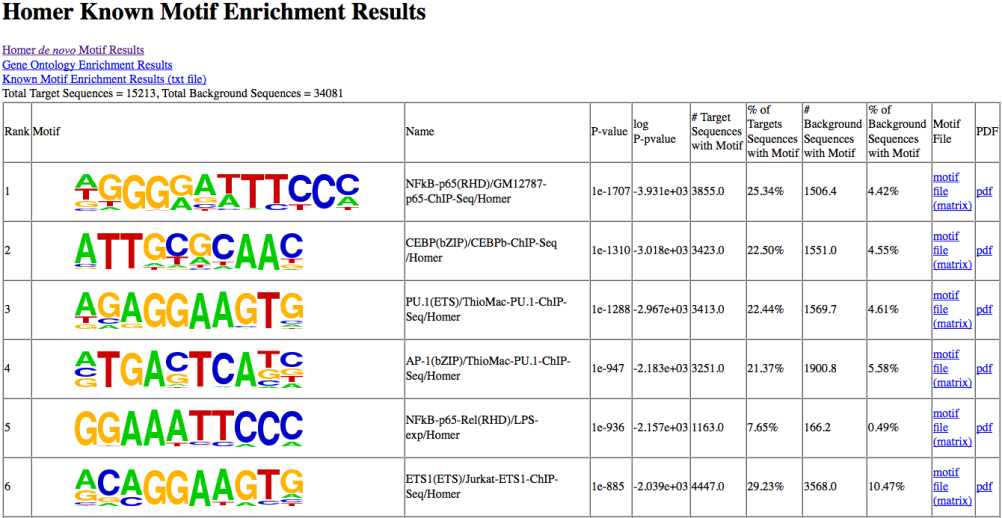
14. 已知motif的输出
该输出也是以网页形式展现,页面结果根据enrichement进行排序,具体如下: