本文希望帮助大家快速使用GSEA软件进行基因集富集分析,如果希望了解GSEA分析原理话,可以看之前的文章使用clusterProfiler包进行富集分析。GSEA软件的使用可以分为以下四个步骤:
数据的准备
- 表达矩阵
- 表型文件
导入数据
运行GSEA
查看结果
下面以GEO芯片数据集(GSE7476)为例,一步步演示GSEA软件的使用。该数据集来自GPL570芯片平台,共有12个样本,包括9个肿瘤膀胱组织和3个正常膀胱组织。GSEA软件版本为4.0.1。
数据准备
表达矩阵
下载GSE7476数据集,如果网络差,可能导致数据下载不全,需要删除已下载的数据,重新下载。
setwd('./task-12')
rm(list = ls())
library(GEOquery)
gset 'GSE7476',destdir = '.', getGPL = T, AnnotGPL = T)
gset1 1]]
expr # 表达矩阵
pdata # 样本信息
fdata # 探针注释信息
筛选探针:
library(tidyverse)
fdata1 %
select(ID, `Gene symbol`) %>%
rename(symbol = `Gene symbol`) %>%
filter(!(symbol == '' | str_detect(symbol, '///')))
ID转换:
expr1 %
as.data.frame() %>%
rownames_to_column('ID') %>%
inner_join(fdata1, by = 'ID') %>%
relocate(symbol, .after = 'ID')
# 多个探针对应同一个symbol,取均值
library(limma)
expr2 1, 2)], ID = expr1$symbol)
分位数标准化:
expr3 = normalizeBetweenArrays(expr2) %>%
as.data.frame()
样本名有点奇怪,换成标准命名(可选操作):
colnames(expr3) 表达矩阵中前3个样本为对照组,这里我们将其放在数据集最后面:
expr4 %
relocate(1:3, .after = last_col())
expr4[1:4, 1:4]
## GSM180994 GSM180995 GSM180996 GSM180997
## CFAP53 3.466569 3.547136 3.213648 3.340319
## ARMCX4 3.823789 3.858049 3.731523 4.013900
## RBBP6 5.281614 5.188695 5.288568 5.143793
## CENPBD1 4.517881 4.237953 4.273378 4.126023
最后将表达矩阵整理成GSEA要求的数据格式:
gsea_expr %
rownames_to_column(var = 'NAME') %>%
mutate(DESCRIPTION = 'na') %>%
relocate(NAME, DESCRIPTION)
gsea_expr[1:4, 1:4]
## NAME DESCRIPTION GSM180994 GSM180995
## 1 CFAP53 na 3.466569 3.547136
## 2 ARMCX4 na 3.823789 3.858049
## 3 RBBP6 na 5.281614 5.188695
## 4 CENPBD1 na 4.517881 4.237953
# 保存表达矩阵
write_delim(gsea_expr, 'gsea_expr.txt',
delim = '\t', col_names = T, na = '')
表型文件
创建表型文件:
group_list num = c('12','2','1',rep(NA, 9)),
group = c('#','tumor','control', rep(NA, 9)),
name = c(rep('tumor', 9), rep('control', 3))
) %>%
t() %>%
as.data.frame()
group_list
## V1 V2 V3 V4 V5 V6
## num 12 2 1
## group # tumor control
## name tumor tumor tumor tumor tumor tumor
write_delim(group_list, 'group_list.cls',
delim = ' ', col_names = F, na = '')
导入数据
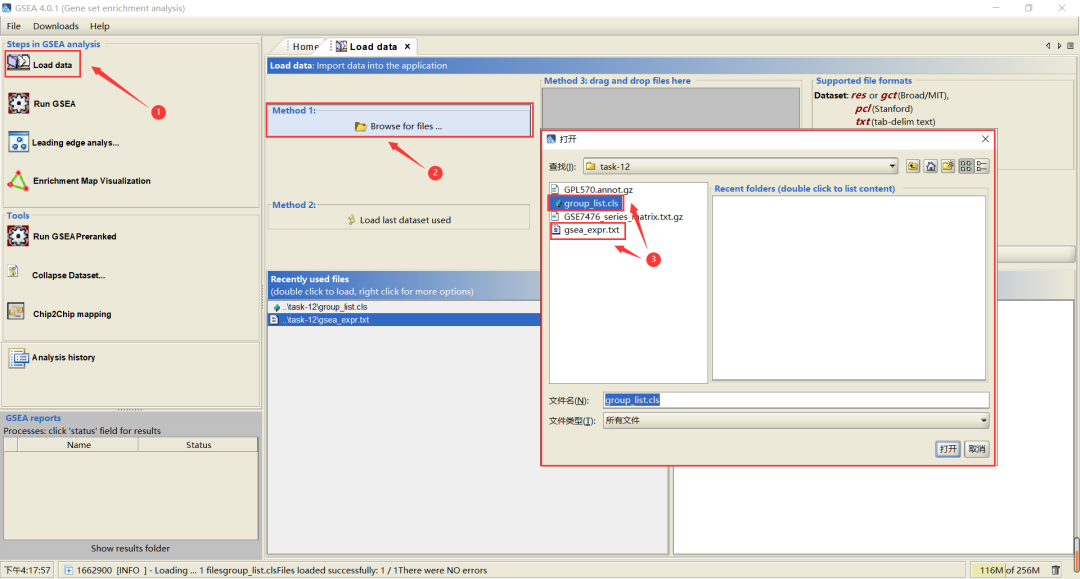
如下图所示,打开软件后,首先点击“load data”,然后选择“Method 1”,找到数据所在文件夹,分别点击“gesa_expr.txt”和“group_list.cls”文件,选择打开,这样数据就上传成功了。
运行GSEA
数据导入之后,点击“Run GSEA”,这里需要我们设置GSEA参数,参数设置包括三个方面:
- 必须参数
- 基本参数
- 高级参数
必须参数
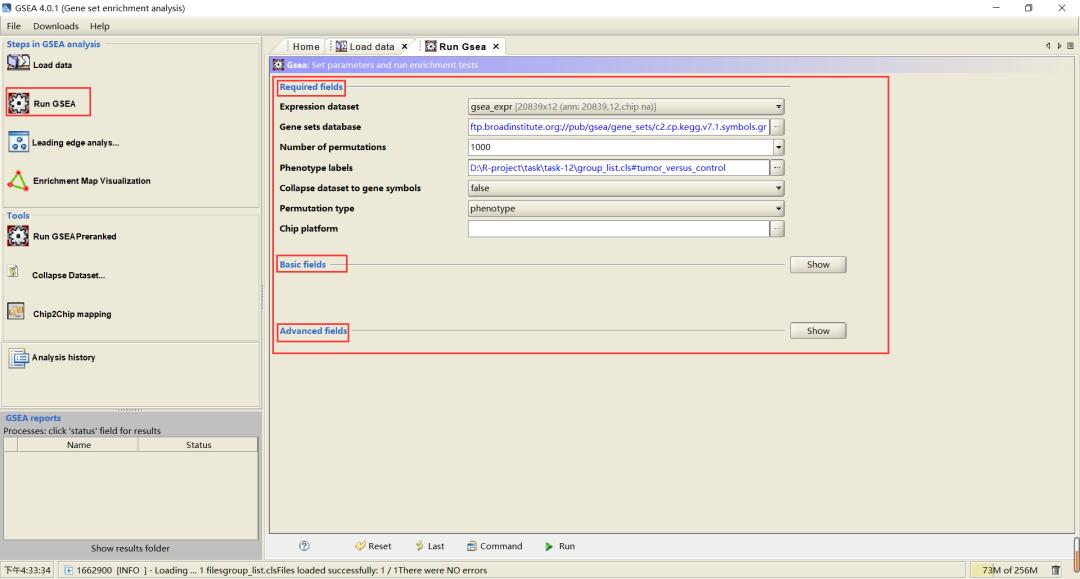
参数设置如上图所示,这里解释下这些参数的作用:
Expression dataset: 使用已上传的表达矩阵“gesa_expr.txt”。
Gene sets database: 选择基因集“c2.cp.kegg.v7.1.symbols.gmt”进行KEGG富集分析。也可以选择其他基因集,如GO基因集。
Number of permutations: 置换检验的次数,一般都设置为1000。
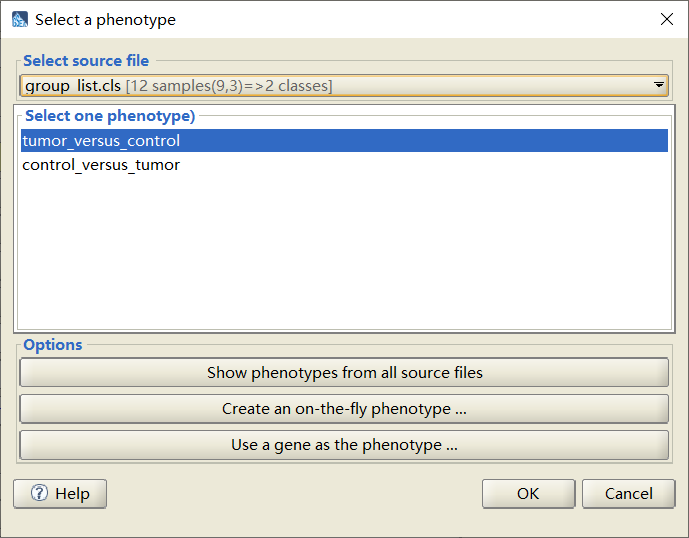
Phenotype labels: 选择表型标签,注意不同组的顺序,需要和表达矩阵一致,这里我们选择“tumor versus control”。
Collapse dataset to gene symbols: 基因名是否转为gene symbol,这里选择“false”,因为我们表达矩阵用的就是gene symbol,不需要再次转换。
permutation type: 置换检验的类型,一般有两种选择,如果样本过少则选择“gene_set”,这里我们选择“pheno type”。
Chip platform: “Collapse dataset to gene symbols ”设置为“false”时,该参数就无需使用。
基本参数
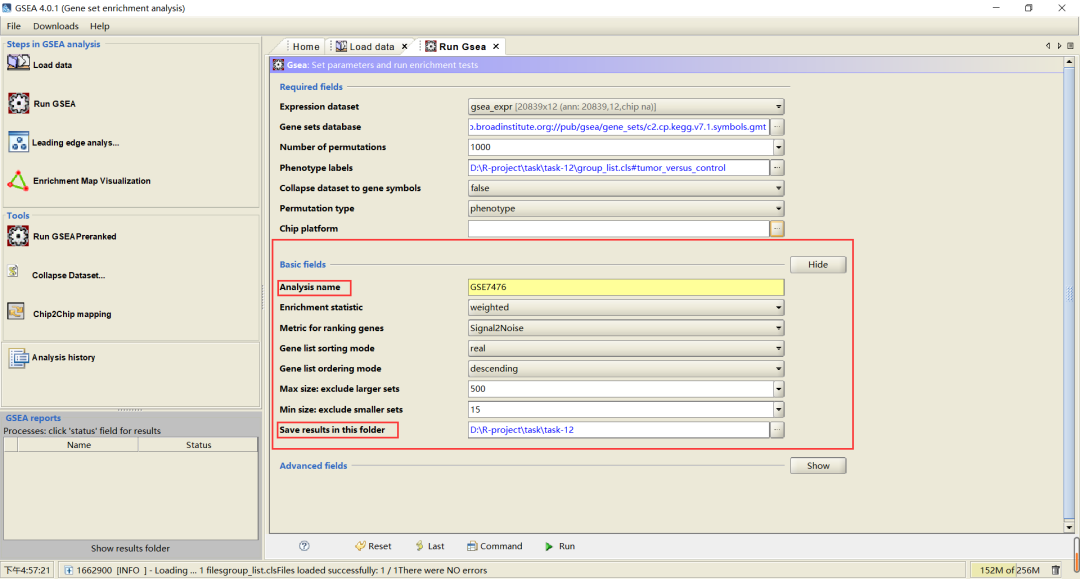
基本参数中,一般只要设置“Analysis name”和“Save results in this folder”参数,其余参数采用默认即可:
- Analysis name: 富集分析结果文件名。
- Save results in this folder: 富集分析结果存储位置。
高级参数
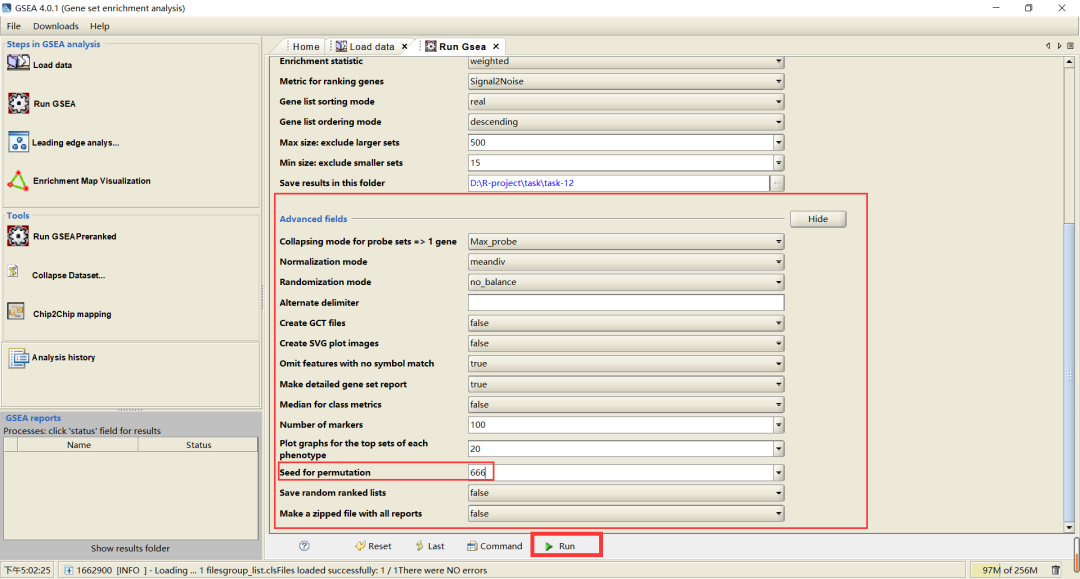
高级参数一般也不用修改,这里我们设置“Seed for permutation”为“666”,是为了运行结果可重复。参数设置完成后,点击“Run”,即可开始运行GSEA。
查看结果
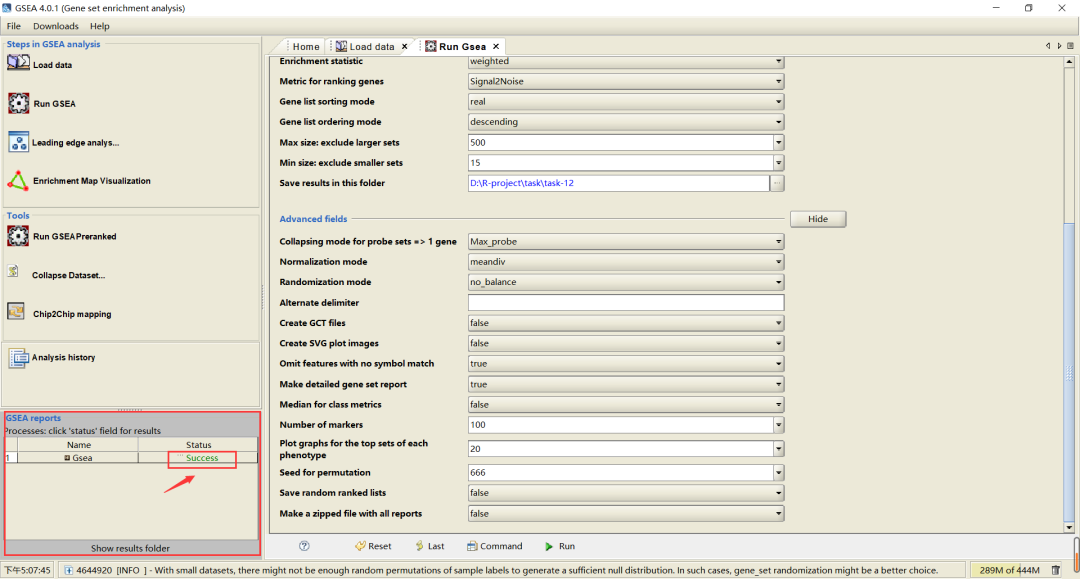
运行结束后,点击“Success”即可进入结果页面查看结果。
结果报告如下图所示:主要分为两部分:
- 在癌症组中基因集富集情况
- 在对照组中基因集富集情况
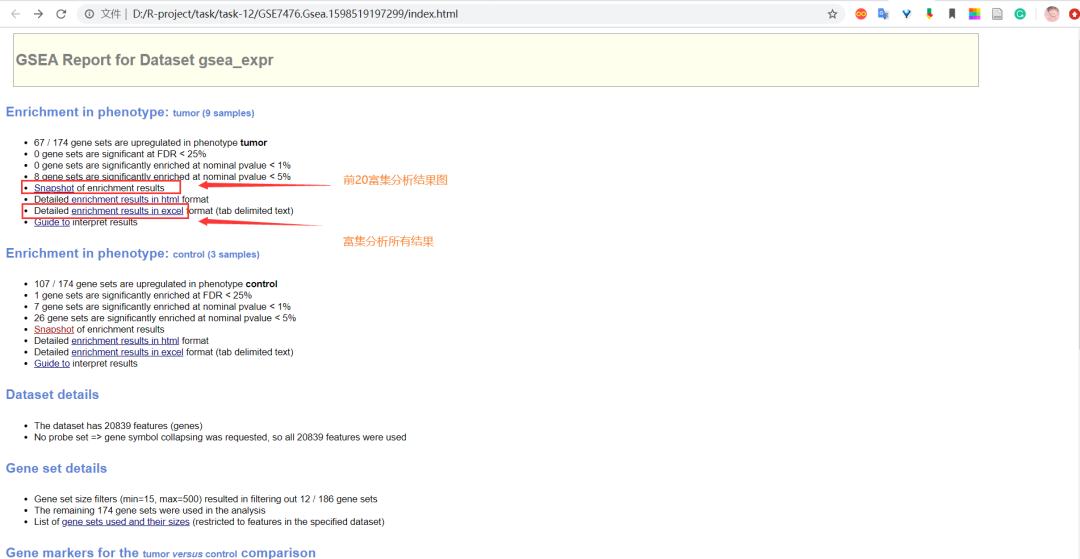
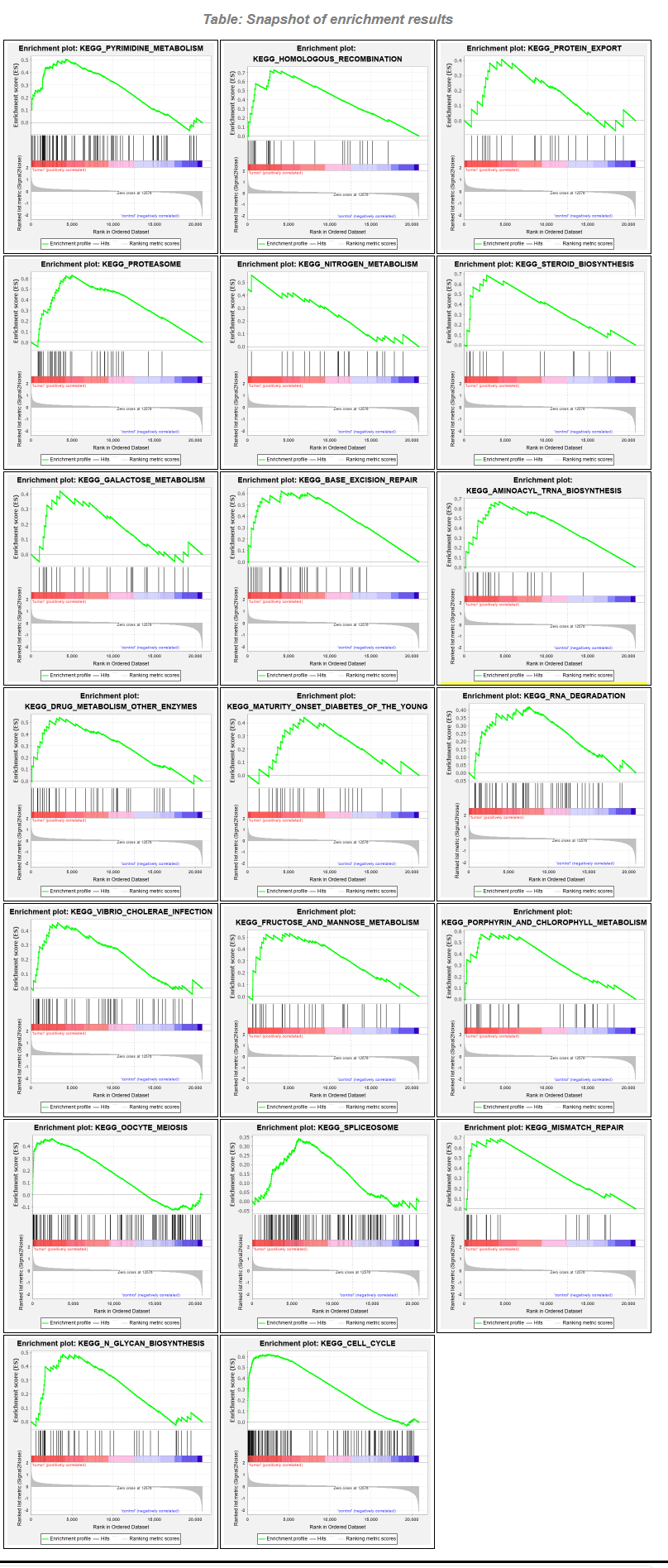
点击“snapshot”查看癌症组前20富集terms:
点击“enrichment results in html”查看所有富集分析结果:
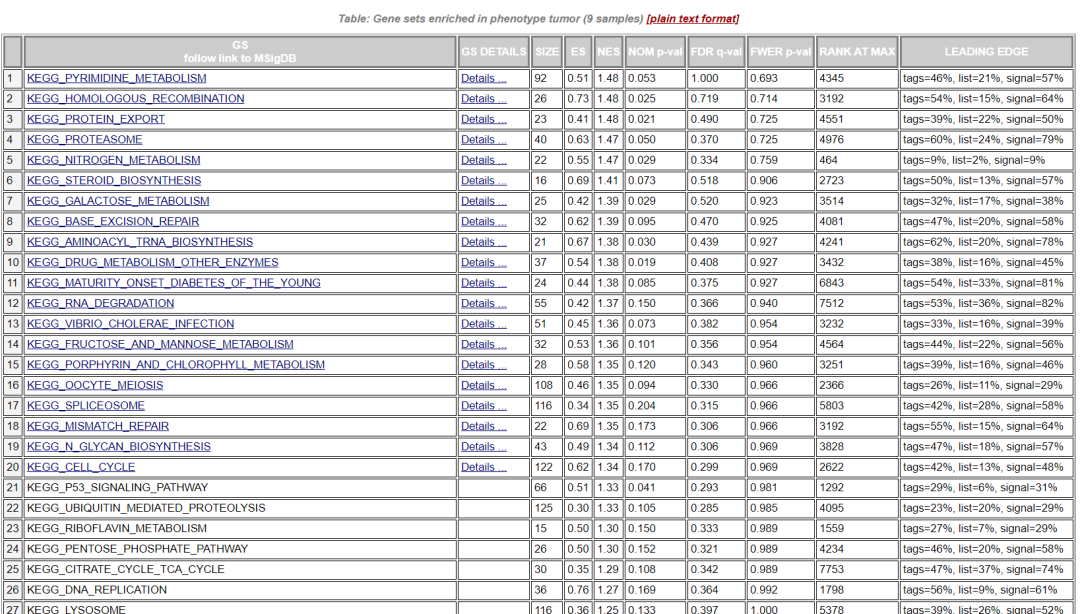
当term满足条件|NES| > 1, NOM p-val < 0.05, FDR q-val < 0.25时,被认为是显著富集。
以上就是使用GSEA对芯片数据集进行基因集富集分析的全部过程了,除了使用芯片数据,也可以使用RNA-seq数据,二者分析流程差不多,这里就不再次介绍了。
GSEA下载地址:https://www.gsea-msigdb.org/gsea/downloads.jsp。
下方是留言小程序入口,点击留言体验
写留言