记录贴—使用cd-hit对orthofinder的准备数据进行预处理
在orthofinder运行后,我又遇到了新的问题—orthofinder的结果文件没有gene_tree文件,为此,我又试了三次,每次都对命令进行一些修改,大约花费了一个月的时间,但结果依旧不如人意,经过一点一点的调整,我发现问题还是出现在fasta数据文件上。
我这三次运行期间,日志文件都有如下警告: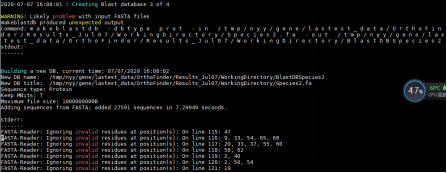
之前我用的去除冗余数据的脚本是从网上搜到的,但我感觉去除冗余的效果不是很好(数据处理前后大小变化不大)(当然可能是因为我看不懂脚本,用错了),然后今天我搜到了cd-hit,具体内容可参考教程 | 如何用cd-hit去除冗余序列?_【快资讯】 https://www.360kuai.com/pc/989e0958f220cf998?cota=4&tj_url=so_rec&sign=360_57c3bbd1&refer_scene=so_1
根据上述教程,我用了如下命令
$ cd-hit -i input.fasta -o output.fasta -c 0.9 -aS 0.9 -d 0
(参数-c 和 -aS,我都设成了0.9,因为我认为这样精度更高一点,如有问题大家可以提出来,我也是个小白 )
根据上述命令,挨个对自己的蛋白数据处理一下,将得到的fasta文件汇总到一个文件夹,然后继续运行orthofinder
$ orthofinder -f data -M msa -S blast -T iqtree -t 10 2>orthofinder.log
orthofinder使用可参考基因家族扩张与收缩分析及物种进化树构建(上) - 简书 https://www.jianshu.com/p/8c6ef557cc71
运行正常的话,应该如下图所示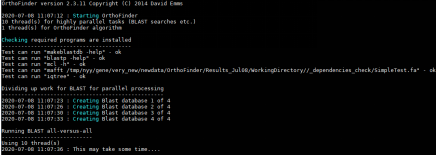
在解决这个问题之前,我也走了很多弯路,也求助了小伙伴,小伙伴提出,会不会是线程(参数t)设置的太大,导致结果文件被砍掉了部分(我开始设置t=24,a=24,后面直接设置成t=10),但是仍旧不对。后来又检查空间是不是满了,我又删掉好多文件,为了省一点地方。这是第四次运行了,目前没有出现警告,希望能够得到tree的文件。欢迎小伙伴一起讨论!
PS:
在对准备的蛋白序列进行处理时,我还考虑过,是不是每行字符过多的问题,为了规整每行字符数(英文字符),用了如下命令:
$ fasta_formatter -i genome.fa -o genome_format.fa -w 70
(fasta_formatter 是 fastx-tookit 的一个命令,可以使用conda install fastx-tookit )
(-i输入文件名 ,-o输出文件名,-w确定每行的字符个数)
使用时,我发现后缀必须是.fa,因为我写成.fasta,运行时会报错
(关于这个后缀问题,大家可以注意一下,也许就是因为后缀不对导致结果运行不了呢,我以前没有注意过这个问题,但我觉得不失为解决问题的一个角度吧)
经过尝试,orthofinder的运行与每行字符数没什么关系,此处仅为记录。
如果orthofinder运行报错,大家不妨关注一下去冗余数据的过程是不是有什么问题。
欢迎大家批评指正!