以下文章来源于简书,作者 biobin,文章已获原作者授权。
前言
关于
clusterProfiler这个 R 包就不介绍了,网红教授宣传得很成功,功能也比较强大,主要是做 GO 和 KEGG 的功能富集及其可视化。简单总结下用法,以后用时可直接找来用。 首先考虑一个问题:
clusterProfiler做 GO 和 KEGG 富集分析的注释信息来自哪里? GO 的注释信息来自 Bioconductor,提供了19个物种的 org 类型的 GO 注释信息,如下表所示。Bioconductor 中更多的注释包可参考下面这个链接,很乱,大多数我都不知道干啥用的。
- http://www.bioconductor.org/packages/release/data/annotation/
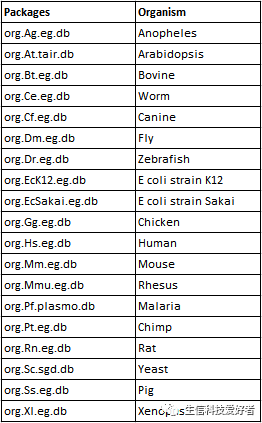
clusterProfiler通过 KEGG 数据库的 API 来获取, https://www.kegg.jp/kegg/rest/keggapi.html。首先是一个物种所有基因对应的 pathway 注释文件,比如人的:http://rest.kegg.jp/link/hsa/pathway。
其次还需要 pathway 的描述信息,比如人的:http://rest.kegg.jp/list/pathway/hsa。
关于 KEGG 数据库全部的物种及其简写(三个字母)如下列表(部分截图):
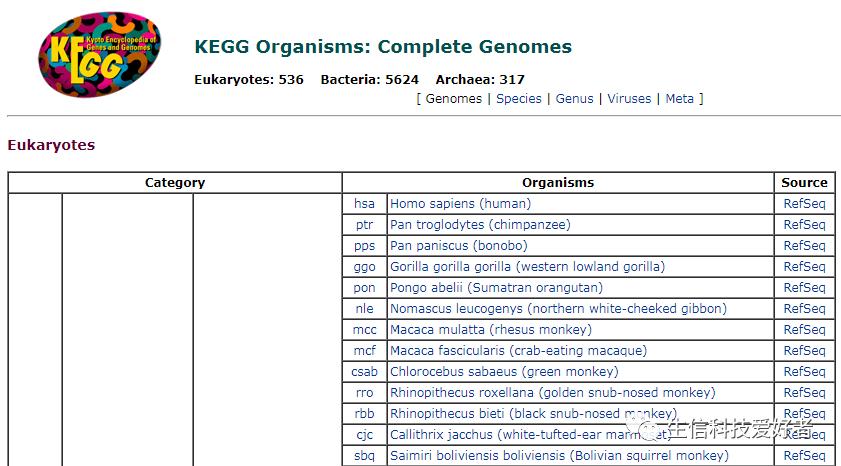
因此对于以上已有 pathway 注释的物种,只需要将物种简写输入给clusterProfiler, 它会通过联网自动获取该物种的 pathway 注释信息。
以上都是有物种信息的情况,那么对于无物种信息的项目怎么办?
GO 可以通过读取外部的 GO 注释文件进行分析。关于基因的 GO 注释,interproscan、eggnog-mapper和blas2go等软件都可以做,不过输出格式有些不同。clusterProfiler需要导入的 GO 注释文件的格式如下:

需要包含以上三列信息,这 3 列信息任意顺序都可。
clusterProfiler包只针对含有 OrgDb 对象,如果是公共数据库中有该物种注释信息,只是未制作成 org.db 数据库(标准注释库),则可以不需要从头注释,只需手动制作 org.db 数据库类型,完成后直接使用即可,代码如下:
source("https://bioconductor.org/biocLite.R")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("AnnotationHub") # 一个包含大量注释信息的数据库,里面有很多物种及来源于很多数据库的注释信息。
BiocManager::install("biomaRt")
library(AnnotationHub)
library(biomaRt)
hub # 建立AnnotationHub对象(视人品,网不行加载不了)
# unique(hub$species) # 查看AnonotationHub里面物种
hub$species[which(hub$species=="Solanum")] # 看AnonotationHub里是否包含想要的物种
# Solanum是番茄的拉丁名
query(hub, "Solanum") # 查看该物种信息
hub[hub$species=="Solanum" & hub$rdataclass == "OrgDb"] # OrgDb属于rdataclass中,因此查看下该物种有没有OrgDb
Solanum.OrgDb "AH59087"]] # AH59087是番茄对应的编号
# 制作为标准注释库,就可和模式生物一样使用了
同样地,对于 pathway 数据库中没有的物种,也支持读取基因的 pathway 注释文件,然后进行分析,注释文件的格式如下:

富集分析
通常用的富集分析有 ORA、FCS 和拓扑三种方法。ORA 简单来说就是超几何检验或 Fisher 精确检验,大同小异,都符合超几何检验,这也是目前用的最多的方法,优劣不谈。FCS 的代表就是 GSEA,即基因集富集分析,优劣亦不谈。clusterProfiler提供了这两种富集分析方法。
1. ORA(Over-Representation Analysis)
GO 富集参考代码:
#标准富集分析
ego gene = gene$entrzID,
keyType = "ENTREZID",
universe = names(geneList), #背景基因集,可省
OrgDb = org.Hs.eg.db,
ont = "CC",
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05,
readable = TRUE)
#通过导入外部注释文件富集分析
data "go_annotation.txt",header = T,sep = "\t")
go2gene 2, 1)]
go2name 2, 3)]
x 一些参数说明:
gene:差异基因对应的向量;
keyType:指定基因ID的类型,默认为 ENTREZID, 可参考
keytypes(org.Hs.eg.db)类型 ;OrgDb:指定该物种对应的 org 包的名字;
ont:代表 GO 的 3 大类别,BP,CC,MF,也可是全部 ALL;
pAdjustMethod:指定多重假设检验矫正的方法,有
"holm","hochberg","hommel","bonferroni","BH","BY","fdr","none"中的一种;cufoff:指定对应的阈值;
readable=TRUE:代表将基因 ID 转换为 gene symbol。
KEGG Pathway 富集参考代码:
#标准富集分析
ego gene = gene,
keyType = "kegg",
organism = 'hsa',
pvalueCutoff = 0.05,
pAdjustMethod = "BH",
qvalueCutoff = 0.05
)#通过外部导入注释文件富集
data "pathway_annotation.txt",header = T,sep = "\t")
go2gene 2, 1)]
go2name 2, 3)]
x 默认基因 ID 为 kegg gene id,也可以是 ncbi-geneid,ncbi-proteinid,uniprot 等。
- organism 物种对应的三字母缩写,其他参数同 GO 富集。
ID 转换函数:
library(clusterProfiler)
bitr_kegg("1",fromType = "kegg",toType = 'ncbi-proteinid',organism='hsa')
library(org.Hs.eg.db)
keytypes(org.Hs.eg.db) #支持的ID类型
bitr(gene, fromType = "ENTREZID", toType = c("ENSEMBL", "SYMBOL"), OrgDb = org.Hs.eg.db)
#以上看出ID转换输入时,可以向量的形式,也可以单列基因名list导入,也可以是内置数据
gene "AASDH","ABCB11","ADAM12","ADAMTS16","ADAMTS18")
gene #字符串
data(geneList, package="DOSE") #富集分析的背景基因集
gene 2]
2. GSEA(Gene Set Enrichment Analysis)
GO 富集参考代码:#标准富集分析
ego geneList = geneList,
OrgDb = org.Hs.eg.db,
ont = "CC",
nPerm = 1000, #置换检验的置换次数
minGSSize = 100,
maxGSSize = 500,
pvalueCutoff = 0.05,
verbose = FALSE)
#通过导入外部注释文件富集分析参考代码:
data "go_annotation.txt",header = T,sep = "\t")
go2gene 2, 1)]
go2name 2, 3)]
x KEGG Pathway 富集参考代码:
#标准富集分析
kk geneList = gene,
keyType = 'kegg',
organism = 'hsa',
nPerm = 1000,
minGSSize = 10,
maxGSSize = 500,
pvalueCutoff = 0.05,
pAdjustMethod = "BH"
)
#通过外部导入注释文件富集
data "pathway_annotation.txt",header = T,sep = "\t")
go2gene 2, 1)]
go2name 2, 3)]
x 可视化
1. GO 富集分析结果可视化
#barplot
barplot(ego, showCategory = 10) #默认展示显著富集的top10个,即p.adjust最小的10个
#dotplot
dotplot(ego, showCategory = 10)
#DAG有向无环图
plotGOgraph(ego) #矩形代表富集到的top10个GO terms, 颜色从黄色过滤到红色,对应p值从大到小。
#igraph布局的DAG
goplot(ego)
#GO terms关系网络图(通过差异基因关联)
emapplot(ego, showCategory = 30)
#GO term与差异基因关系网络图
cnetplot(ego, showCategory = 5)
2. Pathway 富集分析结果可视化
#barplot
barplot(kk, showCategory = 10)
#dotplot
dotplot(kk, showCategory = 10)
#pathway关系网络图(通过差异基因关联)
emapplot(kk, showCategory = 30)
#pathway与差异基因关系网络图
cnetplot(kk, showCategory = 5)
#pathway映射
browseKEGG(kk, "hsa04934") #在pathway通路图上标记富集到的基因,会链接到KEGG官网
 — END—
— END—
 戳原文,更有料!
戳原文,更有料!