
点击上方蓝字关注我们
RNA-seq的建库及分析1RNA-seq的应用
RNA-seq是目前所有高通量测序中应用最广的一种测序方法,可以应用于一下几个方面;
在各种比较情况下(比如正常组织与肿瘤组织之间),所有基因的表达差异变化
mRNA表达亮的差异
mRNA结构上的差异(可变剪接
融合基因、点突变(SNP)
2RNA-seq的方法
在检测mRNA的过程中,我们首先需要解决的问题就是mRNA纯度的问题,换句话说,我们要去除总RNA中的rRNA,因为rRNA占提取的总RNA量的95%以上。

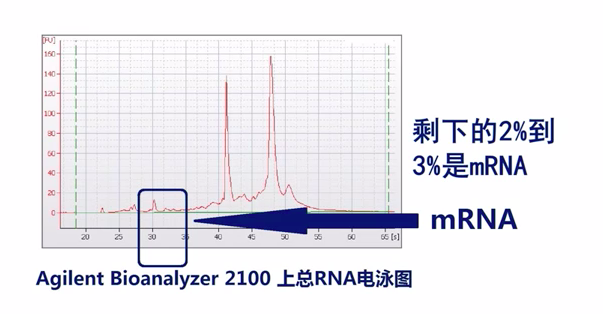
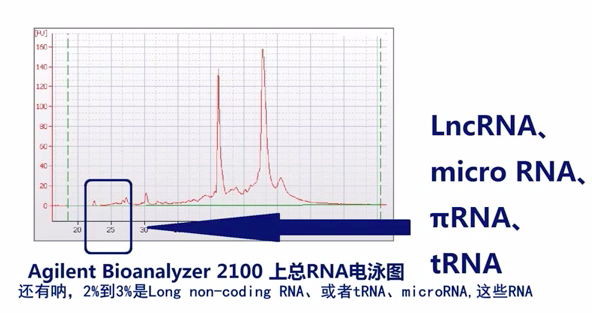
所以,从上图中我们可以看出,如果我们拿总的RNA去测序,我们测到的数据大部分是rRNA。然而,我们知道的是,rRNA在我们人体中是高度保守的,也就是无论你是否患有某种疾病,rRNA的测序结果都是一样的,这样就对我们科研人员来说测序结果将毫无意义!而mRNA是所有RNA中信心含量最丰富的那一部分。因此,我们在测序之前首先要做的就是纯化mRNA,再进行建库测序。
3rRNA去除以及建库目前最常用的就是Illumina公司的Truseq RNA建库方法。
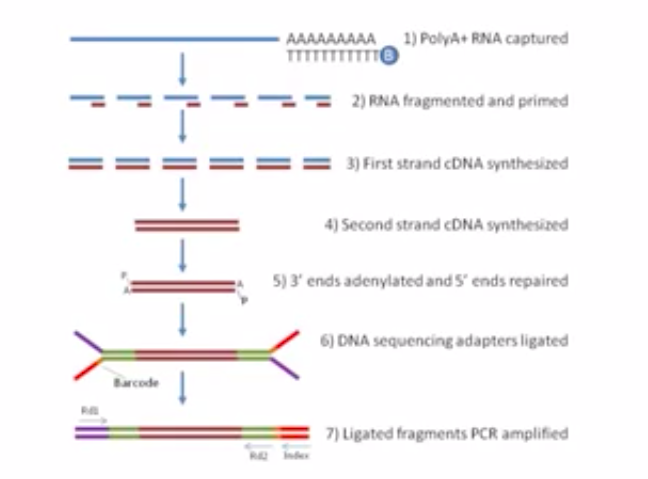
这是建库的流程图。
首先,利用高等生物mRNA都带有Poly(A)尾巴的特点,用带Poly(T)的探针磁珠与总RNA进行杂交,吸附其中的带Poly(A)尾巴的mRNA。
接下来进行磁珠的回收,然后把这些Poly(A)的mRNA从磁珠上洗脱下来。
然后在把洗脱下来的mRNA用镁离子溶液进行处理(镁离子溶液会把mRNA打断)
紧接着,被打断的mRNA片段再用随机引物(dNTP)进行逆转录。逆转录成cDNA(第一条链)后再合成第二条链。这样mRNA就变成了双链的cDNA。
最后,在双链cDNA的两端接上"Y"型的接头,经过PCR扩增,就成为了可以上机测序的文库。我们就可以去Hi Sqe测序仪上进行RNA测序。
注意⚠️,在建库的时候会对mRNA的完整度有较高的要求。只有在完整度高的mRNA中测序才会产生比较好的测序结果。原因就是,我们所用的Poly(T)磁珠只会去吸附靠近3'的Poly(A)序列,如果mRNA发生了降解,也就是mRNA断掉了,那么5'端的mRNA就不会被吸附上了,会在富集的过程中被洗脱掉。由此导致我们的测序结果并不准确。
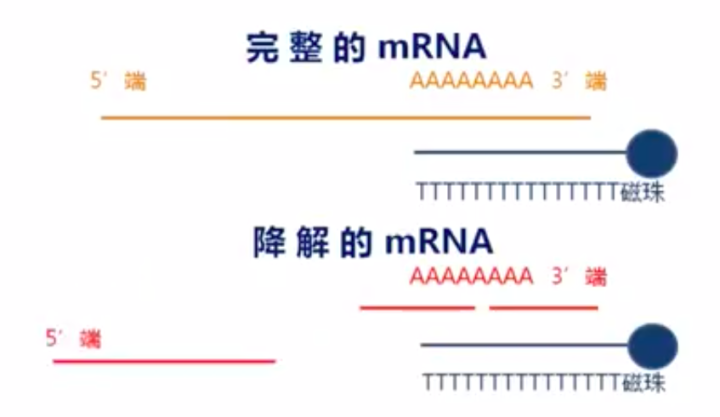
具体的mRNA完整性检测建议大家参考illumina公司的mRNA质量检测。
4RNA-seq的结果分析1、火山图
作为一种针对全转录组的分析,我们希望看到的是一个整体样本的基因表达差异变化,而不仅仅是看少数几个基因的表达差异。为此,科学家研究了一种叫做“火山图”的图形。来比较形象的说明两个样本之间的表达差异。
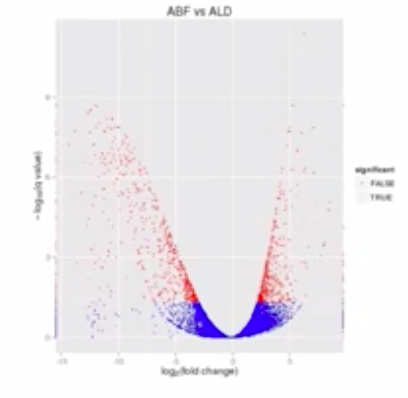
这个图表达的是两个样本之间的表达差异(比如正常组织和癌变组织)横轴表达某个基因是上升了还是下降了,纵轴表述这种差异的置信程度。里面的每一个点表示两个样本中同一个基因的mRNA表达量的变化。
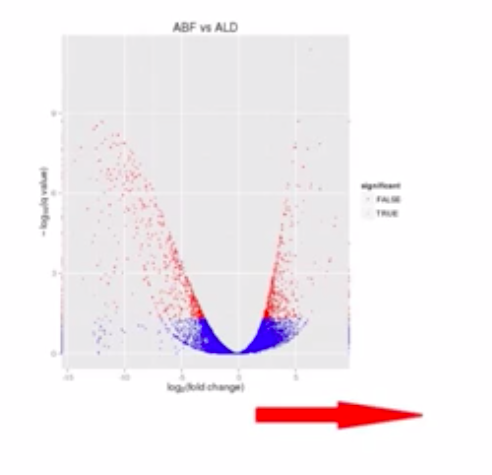
如果这个基因的表达是上调的,那么这个点就是往右移动。相反的,如果这个基因的表达量是下调的,那么这个点就往原点的方向移动。
纵轴表示差异基因的表达的置信程度,如果置信程度越高,那么这个点的纵轴位置就越高
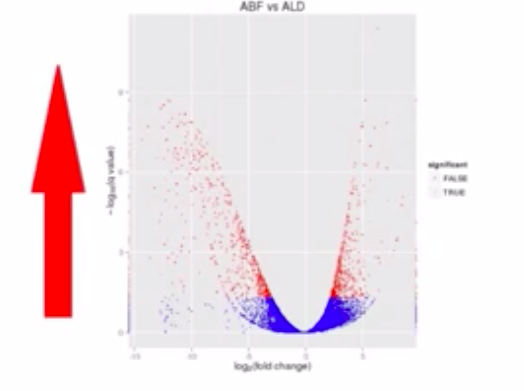
我们在图上做一条水平线,超过这个水平线以上的点的差异水平的置信程度就越高。就把它标为红色。
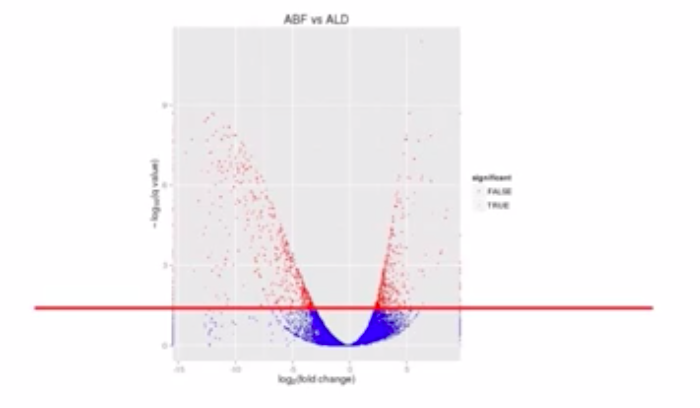
什么差异置信程度?
我们举个例子,A基因在正常组织样本中测到了3200条,而在癌组织样本中测到了100条;B基因在正常组织样本中测到了320条,而在癌组织样本中测到了10条。同样是差了31倍,但由于A基因的样本统计数远远的大于B基因的样本统计数,所以,A基因的差异置信程度会比B基因的差异置信程度高的多。
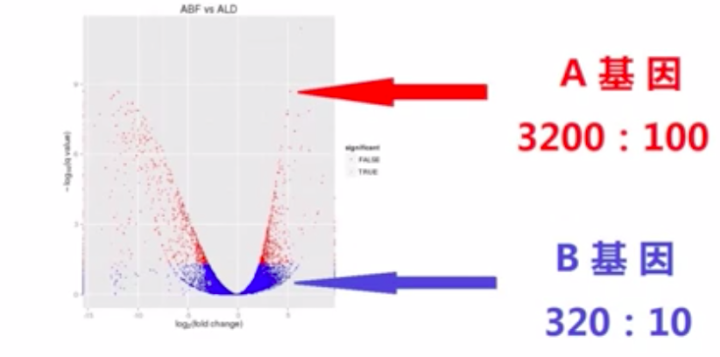
这样我们就可以更加直观的发觉,在置信程度高的图中,会有更多的基因表现出明显的差异。
2、RNA聚类分析
聚类分析是RNA分析中非常常用的一个手段,它是通过多个样本的全基因表达谱的对比来找到它们之间的相似性和相近的关系。

这是一张RNA聚类分析的图,横轴表示样本,纵轴表示基因。通过聚类分析,我们可以看到,在这个群体中,样本被分成了3个群体。每个群体的内部都有相似的表达特征,同时,我们也可以看到,基因的表达是成簇的。
聚类分析有很多的应用,比如我们可以分析疾病的亚型,还可以通过对多个基因在特定疾病当中的表达倾向性来找出可能的、新的、诊断用的Biomaker。
3、GO分析(Gene Ontology分析)
GO分析是一个国际化的、基因功能分类体系,这个体系用一整套动态更新的标准词汇、严格的定义的概念来全面的概括任何生物中基因和基因产物的属性。
GO主要描述基因的三个属性:1、基因参与的生物学过程2、基因的产物的功能3、基因产物在细胞内的空间定位。
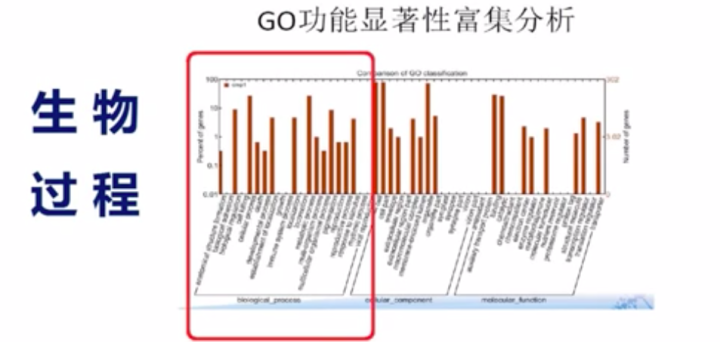
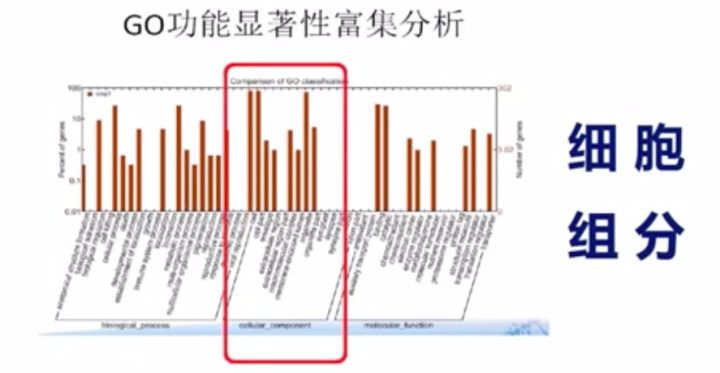

GO分析可以反映上述三个属性中的差异基因的个数及分布的情况
4、KEGG(京都基因和基因组百科全书)通路分析
通路分析:通路是指在系统水平上完成生物的某一功能的基本单位或者局部子网络。
KEGG是目前公认的、最权威的基因功能数据库。其中的通路分析是KEGG的核心内容。目前对于通路的分析、注释,大多数是基于KEGG通路来做的。
散点图是KEGG富集分析结果的图形化的展示方式。在散点图中,KRGG富集程度通过Rich factor、 Qvalue和富集到此通路上的基因个数来衡量。
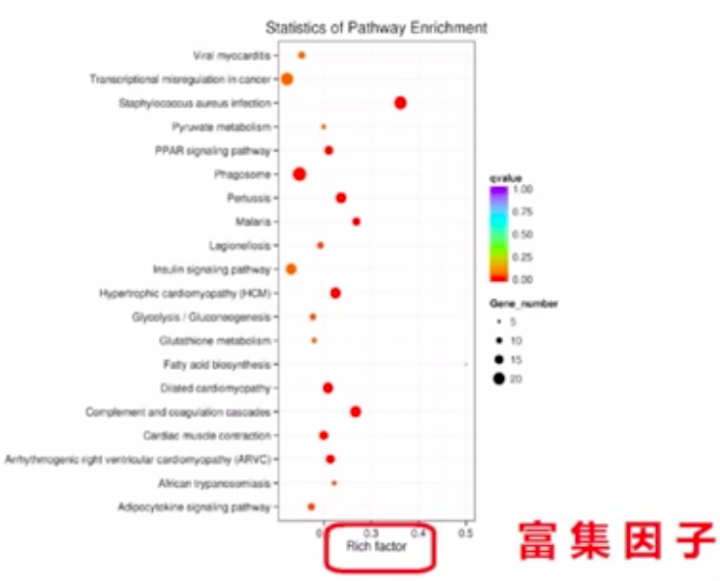
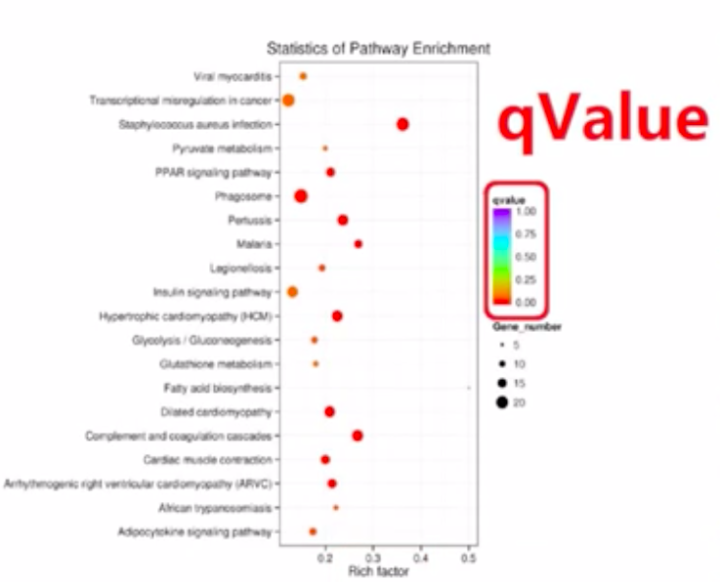
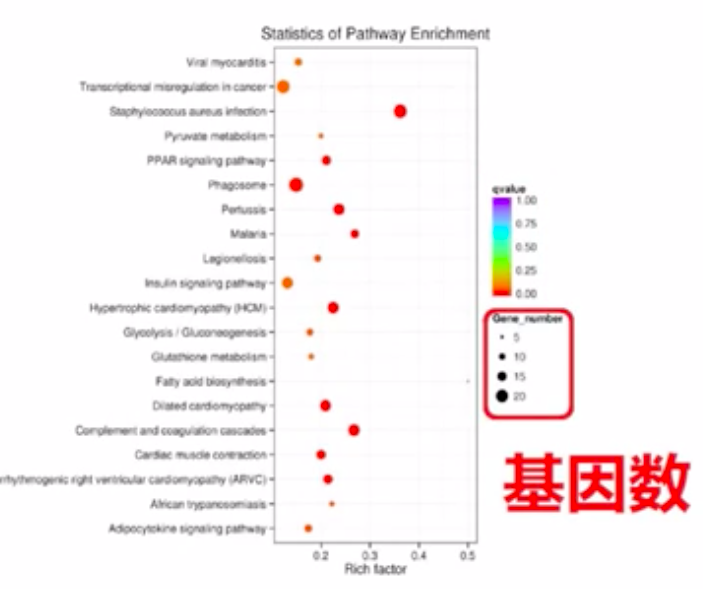
上述图中,点的面积越大,则富集的基因鼠就越多。富集因子越大,则表示富集的程度越大。qvalue是校正之后的pvalue,其值越接近0表示富集程度越显著。
5RNA的结构变异前面所述的都是基于RNA表达量的差异分析,接下来我们要说到的就是在RNA-seq中可以检测到的mRNA上的各种结构上的变异。
所谓结构上饿变异,就是RNA序列的变异。主要包括3种:1、可变剪接 2、融合基因 3、点突变(SNP)
注意:要想测RNA结构变异就必须测序的深度要比较深,一般要测10G的数据量!!!
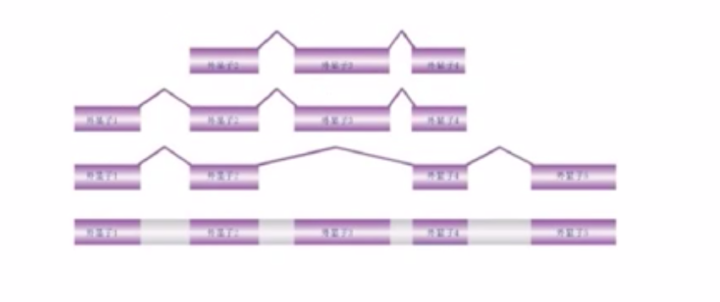
1、可变剪接
可变剪接在真核生物中普遍存在,一般一个人的组织样本中,可以通过高通量测序发现5000个到20000个左右的可变剪接。
2、RNA-seq找融合基因
融合基因是指在原来的基因组上分开的2个基因,因为某种基因染色体发生了重排,重排的结果是让A基因的头接到了B基因的身体上,这样就产生了融合基因。
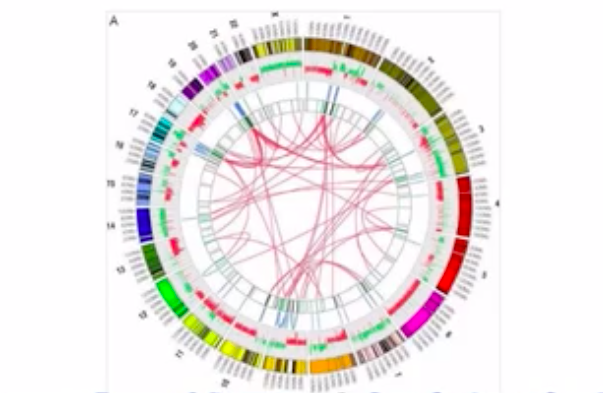
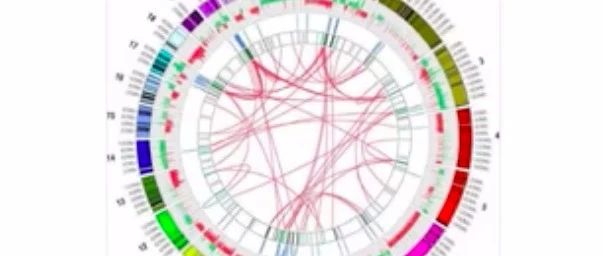
这是一张癌细胞中的融合基因的示意图,
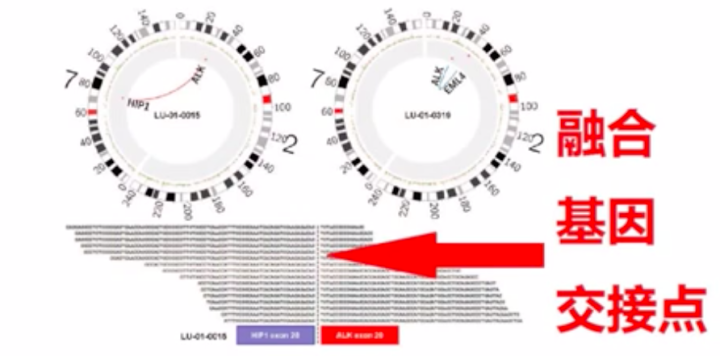
这张图是高通量测序测到的融合基因,我们可以看到,有10几个reads都横跨在这个融合基因的交接点的两侧,由此证明,在这个癌细胞中有这么一个融合基因。
3、RNA-seq找点突变

我们用泡泡图来表示找到的点突变,发生频率最高的基因就用最大的泡泡来表示。频率低的就画小一点的泡泡。整个泡泡呈逆时针排列。
 END▼往期精彩回顾▼瞬间爆红的冰皮月饼—何为冰皮…………COCO新品测评:你长胖不是奶茶的锅!COCO新品测评:你长胖不是奶茶的锅
END▼往期精彩回顾▼瞬间爆红的冰皮月饼—何为冰皮…………COCO新品测评:你长胖不是奶茶的锅!COCO新品测评:你长胖不是奶茶的锅
 微信号:zxy136179484新浪微博:uhuh拒绝吸烟-扫码关注我-
微信号:zxy136179484新浪微博:uhuh拒绝吸烟-扫码关注我-
温馨提示
如果你喜欢本文,请分享到朋友圈,想要获得更多信息,请关注我。