蛋白质是生命活动的体现者,其结构决定着功能。由线性氨基酸组成的蛋白质需要折叠成特定的空间结构才具有相应的生理活性和生物学功能。
解析蛋白质的空间结构对于认识蛋白质的功能、功能的执行、生物大分子间的相互作用,以及医学和药学的发展(如药物靶点的设计等)具有重要意义。为了更快速地了解蛋白质功能,不能只等待蛋白质的测定结果,尤其是对未知蛋白质开展研究之前,通过对蛋白质结构进行预测具有明显的优势。
理论基础
蛋白质的高级结构由其一级结构序列决定,蛋白质可以自发地折叠成它们的天然结构,特别是结构简单的蛋白质小分子。
- 序列相似的蛋白质具有相似的三维结构;
- 不同的蛋白质中相同的结构域(domain)具有相似的功能。
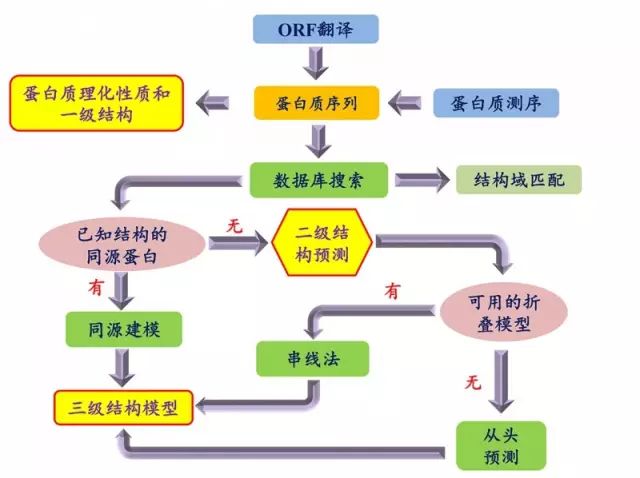 蛋白质结构预测流程 蛋白质理化性质和一级结构分析 1. 分析蛋白质的 pI、Mw、氨基酸组成、消光系数、稳定系数等 (1)进入 Expasy 主页:
蛋白质结构预测流程 蛋白质理化性质和一级结构分析 1. 分析蛋白质的 pI、Mw、氨基酸组成、消光系数、稳定系数等 (1)进入 Expasy 主页:http://web.expasy.org/protparam/
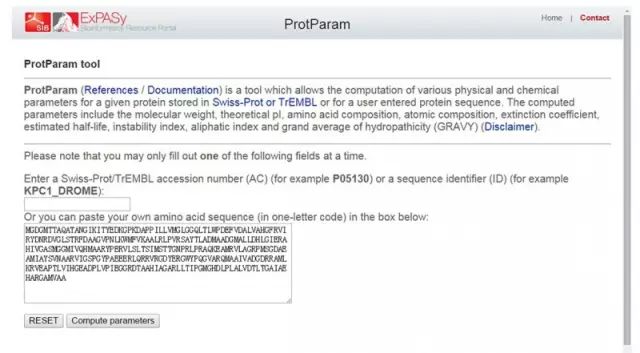 (2)点击 Resource A…Z (3)查找「ProtParam」(protein physical and chemical parameters) (4)粘贴序列进行分析。
(2)点击 Resource A…Z (3)查找「ProtParam」(protein physical and chemical parameters) (4)粘贴序列进行分析。 2. 分析蛋白质的亲水性和疏水性 (1)进入 Expasy 主页: http://web.expasy.org/protscale/ (2)点击 Resource A…Z (3)查找「ProtScale」(protein profile computation and representation) (4)粘贴序列,选择分析方法,进行分析。 (5)蛋白质的亲水和疏水性分析结果,图形显示(亲水用负值表示,疏水用正值表示)。 3. 分析蛋白质的跨膜区 (1)Tmpred 法分析蛋白质的跨膜区: 基于对 Tmbase 数据库的统计分析来预测蛋白质跨膜区和跨膜方向。 http://www.ch.embnet.org/software/TMPRED_form.html
2. 分析蛋白质的亲水性和疏水性 (1)进入 Expasy 主页: http://web.expasy.org/protscale/ (2)点击 Resource A…Z (3)查找「ProtScale」(protein profile computation and representation) (4)粘贴序列,选择分析方法,进行分析。 (5)蛋白质的亲水和疏水性分析结果,图形显示(亲水用负值表示,疏水用正值表示)。 3. 分析蛋白质的跨膜区 (1)Tmpred 法分析蛋白质的跨膜区: 基于对 Tmbase 数据库的统计分析来预测蛋白质跨膜区和跨膜方向。 http://www.ch.embnet.org/software/TMPRED_form.html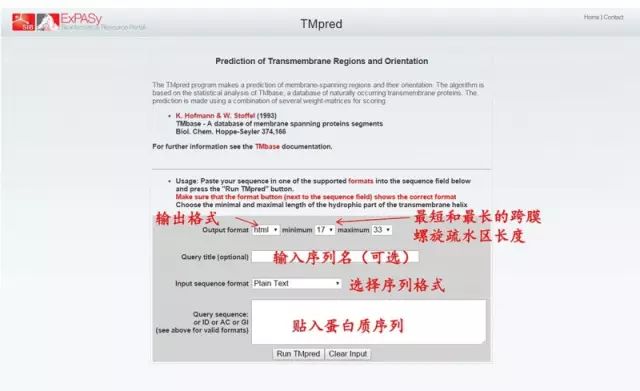
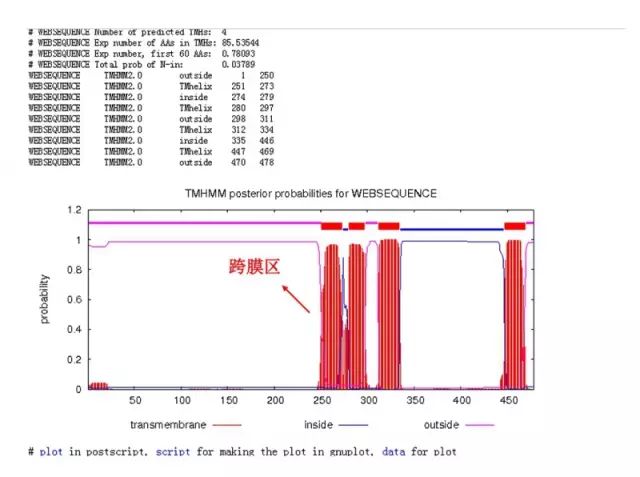
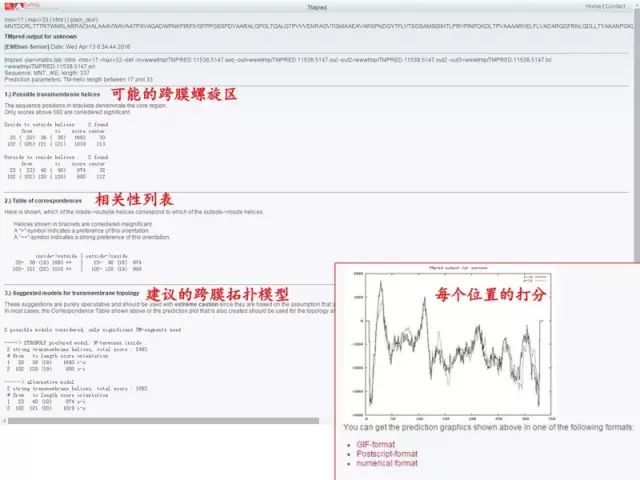 (2)TMHMM 法分析蛋白质的跨膜区: 基于 HMM 方法的蛋白质跨膜区预测工具。 http://www.cbs.dtu.dk/services/TMHMM/
(2)TMHMM 法分析蛋白质的跨膜区: 基于 HMM 方法的蛋白质跨膜区预测工具。 http://www.cbs.dtu.dk/services/TMHMM/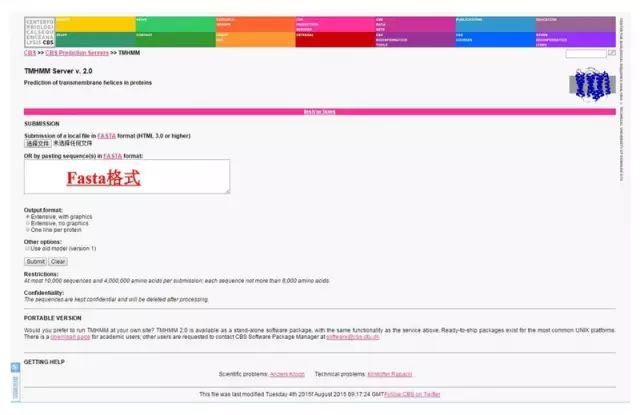
 蛋白质二级结构预测 1. CFSSP http://cho-fas.sourceforge.net/
蛋白质二级结构预测 1. CFSSP http://cho-fas.sourceforge.net/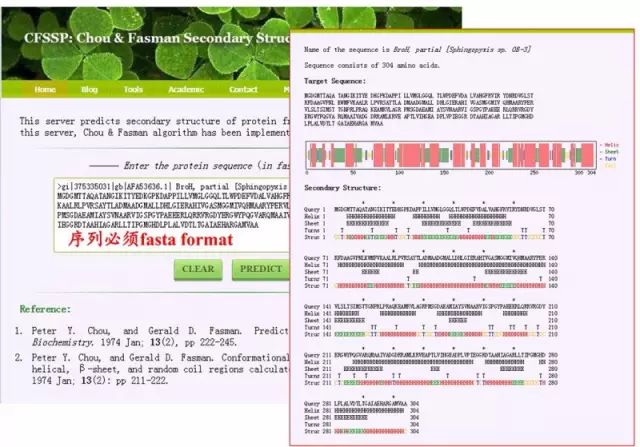 2. PSIPRED http://bioinf.cs.ucl.ac.uk/psipred/
2. PSIPRED http://bioinf.cs.ucl.ac.uk/psipred/
 蛋白质结构域预测 1. InterPro http://www.ebi.ac.uk/interpro/scan.html
蛋白质结构域预测 1. InterPro http://www.ebi.ac.uk/interpro/scan.html
 2. Pfam http://pfam.xfam.org/
2. Pfam http://pfam.xfam.org/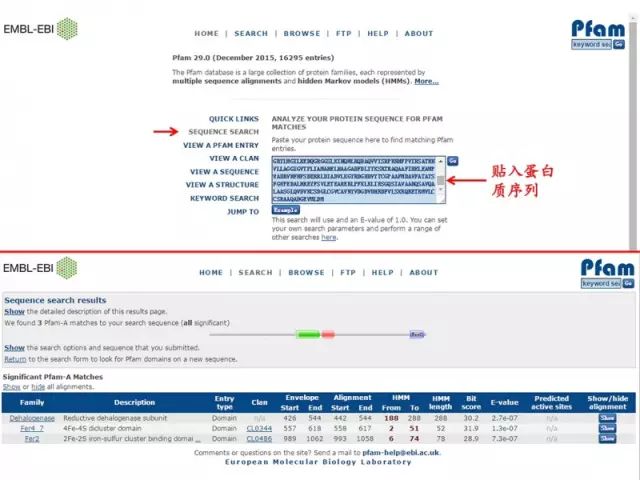 蛋白质三级结构预测 1. 同源建模(SWISS-MODEL) 蛋白质结构同源模建(又称同源模拟、同源建模)的理论基础是蛋白质的三级结构比蛋白质的一级结构更为保守。
蛋白质三级结构预测 1. 同源建模(SWISS-MODEL) 蛋白质结构同源模建(又称同源模拟、同源建模)的理论基础是蛋白质的三级结构比蛋白质的一级结构更为保守。
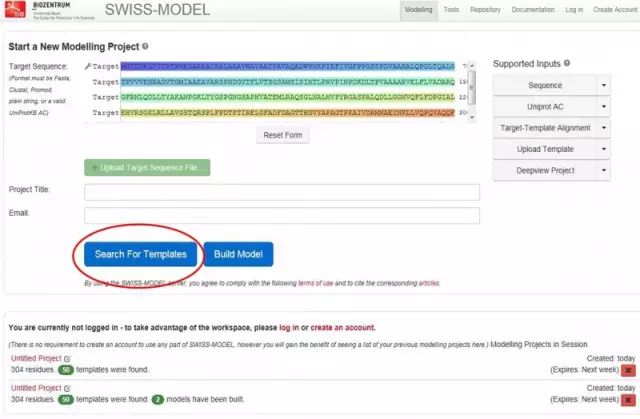
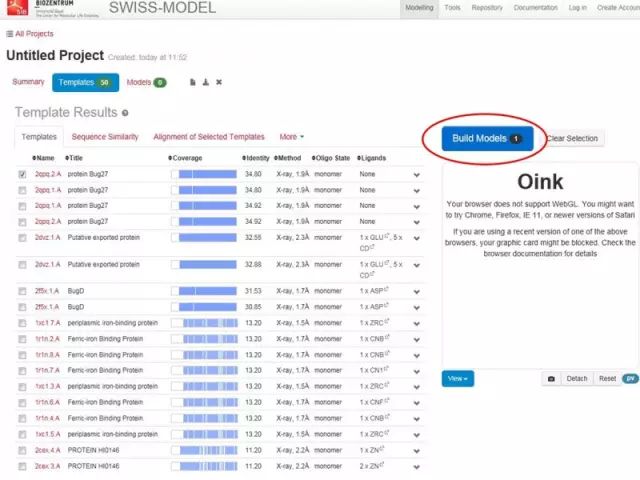
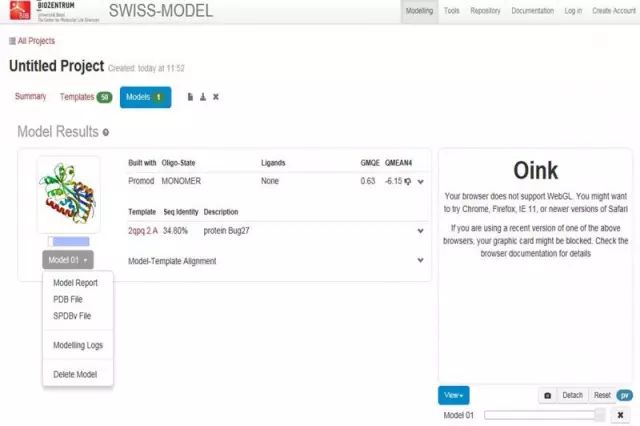


 软件 2:Phyre2 http://www.sbg.bio.ic.ac.uk/phyre2
软件 2:Phyre2 http://www.sbg.bio.ic.ac.uk/phyre2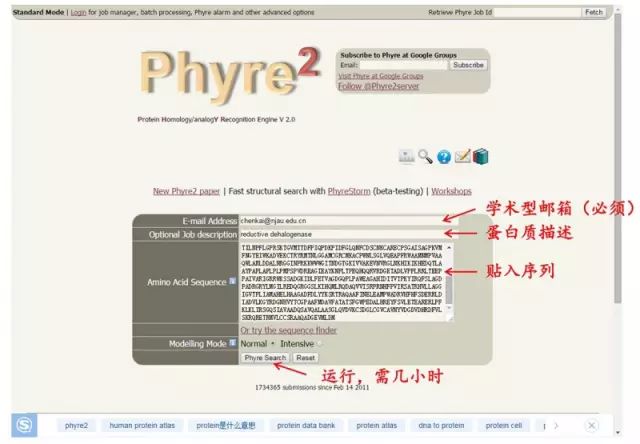 3. 从头计算法 原理: 蛋白质的天然构象对应其能量最低的构象 (热力学理论),因此通过构造合适的能量函数及优化方法,实现从蛋白质序列直接预测其三维结构的目的。 软件:I-TASSER http://zhanglab.ccmb.med.umich.edu/I-TASSER/
3. 从头计算法 原理: 蛋白质的天然构象对应其能量最低的构象 (热力学理论),因此通过构造合适的能量函数及优化方法,实现从蛋白质序列直接预测其三维结构的目的。 软件:I-TASSER http://zhanglab.ccmb.med.umich.edu/I-TASSER/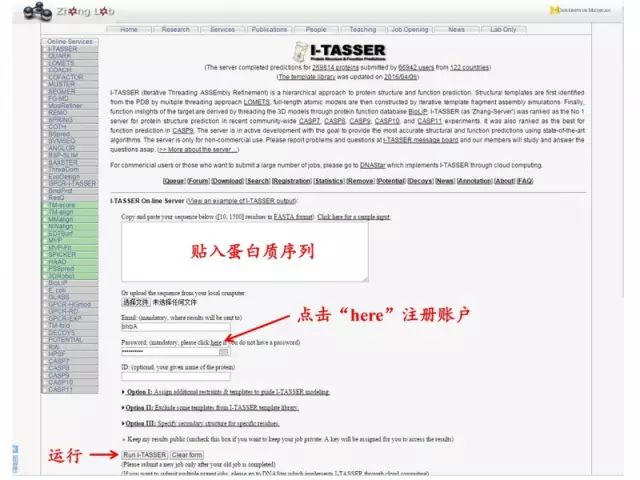 一周左右,结果将发到注册邮箱,进入邮箱打开连接,下载结果。
一周左右,结果将发到注册邮箱,进入邮箱打开连接,下载结果。 XX.pdp 文件用PyMOL 软件打开: http://pan.baidu.com/s/1kUXOhIf
XX.pdp 文件用PyMOL 软件打开: http://pan.baidu.com/s/1kUXOhIf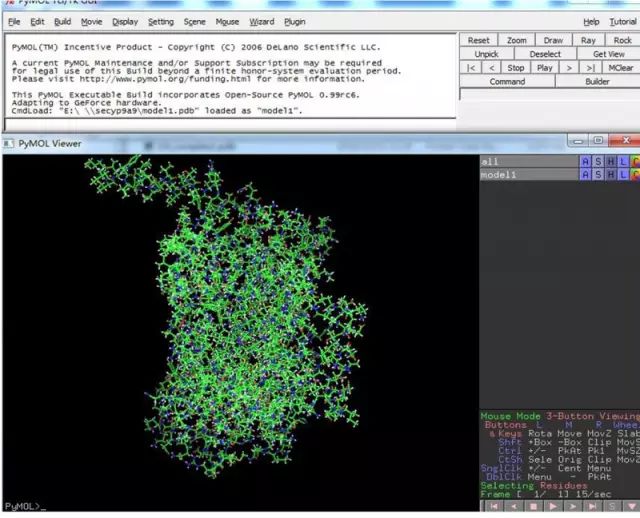
封面来源:站酷海洛 Plus
图片来源:作者提供

版权声明:本文为weixin_42385265原创文章,遵循CC 4.0 BY-SA版权协议,转载请附上原文出处链接和本声明。