Affymetrix芯片原始数据的R语言处理过程网络中很多,为什么我还要写?这要回归到自己做知乎的初衷:1.这是我对自己学习过程的一种总结方式;2.分享自己的所学给需要的各位小伙伴,同时和其他相关文章联系一起,实现生信分析的系统化、完整话。OK,回归正题。
一:原始数据下载
##GEOquery包,此处以数据集GSE3910为例,如有雷同,纯属巧合
####用getGEOSUppFiles()函数获取原始数据#####
library(GEOquery)
GSEName=“GSE3910”
rawdata = getGEOSuppFiles(GSEName)##因网速问题,本人手动下载了该数据集的前6个样本数据并用于后面的分析,正常情况下使用上述函数下载数据后需要解压成单个CEL格式文件,并存放在同一个文件夹中,以备后面使用二:原始数据读入
##2.1 原始数据读取----ReadAffy()函数
##使用choose.dir()函数选择文件夹
dir <- choose.dir(caption = "Select folder")
# 列出CEL文件,保存到变量
cel.files <- list.files(path = dir, pattern = ".+.cel.gz$", ignore.case = TRUE,
full.names = TRUE, recursive = TRUE)
# 查看文件名
basename(cel.files)
data.raw <- ReadAffy(filenames = cel.files)#读入数据
##读入芯片的默认样品名称是文件名,用sampleNames函数查看并修改:
sampleNames(data.raw)##
##2.2 样本重命名--使用stri_sub函数删除CEL等,仅保留GSEM号
library(stringi)
sampleNames(data.raw)<-stri_sub(sampleNames(data.raw),1,9)##8或9或10
sampleNames(data.raw)##
##2.3 构建样本分组信息
pData(data.raw)
group_file=pData(data.raw)
group_file$sample=rownames(group_file)
group_file$disease=rep(c("diagnosis","relapse"),time=3)
group_file#查看分组信息三:基因芯片数据预处理---质量控制--查找并删除芯片质量差的芯片
#质量控制包括:质量分析报告、RLE箱线图、NUSE箱线图、RNA降解图,通过上述分析,进而剔除质量不合格的样本;
library(hgu133plus2cdf)
library(hgu133plus2.db)
#PM和MM查看:
# Perfect-match probes
pm.data <- pm(data.raw)
head(pm.data)
# Mis-match probes
mm.data <- mm(data.raw)
head(mm.data)3.1 灰度图 显示芯片扫描图像的灰度,芯片图像中有斑块详细的可能就是质量差的芯片
library(affy)
##绘制所有图片并保存
for (i in 1:length(sampleNames(data.raw))){
name = paste("image",sampleNames(data.raw)[i],".jpg",sep="")
jpeg(name)
image(data.raw[,i])
dev.off()
}
#此处及后续只显示第一张,其他略
##3.2 Chip pseudo-images 根据探针水平模型的权重或是残差,就可以生成对应的pseudo-images
##3.2.1 拟合探针水平模型 以“加权最小二乘法”对数据集进行回归计算
library(affyPLM)
Pset <- fitPLM(data.raw, output.param = list(varcov="none"))
##3.2.2 生成权重图
image(Pset, type = "weights",which = 1)
for (i in 1:length(sampleNames(data.raw))){
name = paste("pseudoimageweights", sampleNames(data.raw)[i], ".jpg", sep = "")
jpeg(name)
image(Pset, type = "weights",which = i)
dev.off()
}
##3.2.3 生成残差图
image(Pset, type = "resids",which = 1)
for (i in 1:length(sampleNames(data.raw))){
name = paste("pseudoimageresids", sampleNames(data.raw)[i], ".jpg", sep = "")
jpeg(name)
image(Pset, type = "resids",which = i)
dev.off()
} 
##3.2.4 生成RLE,NUSE图
#生成RLE图
library(affyPLM)
RLE(Pset)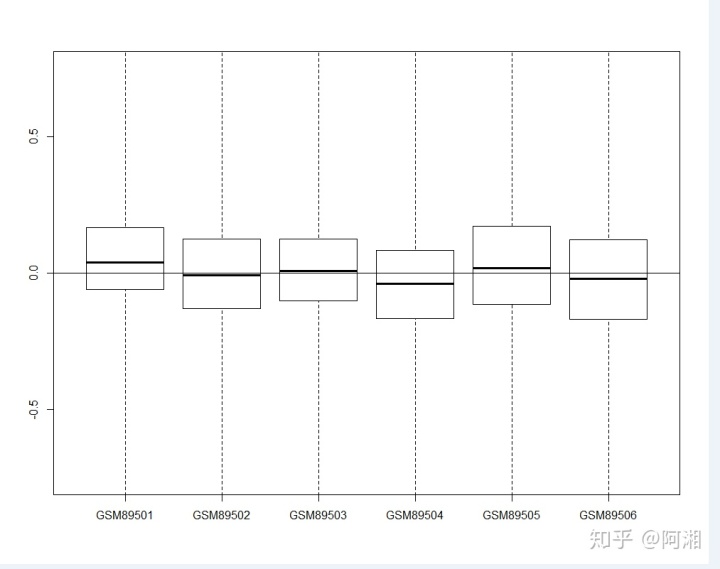
NUSE(Pset)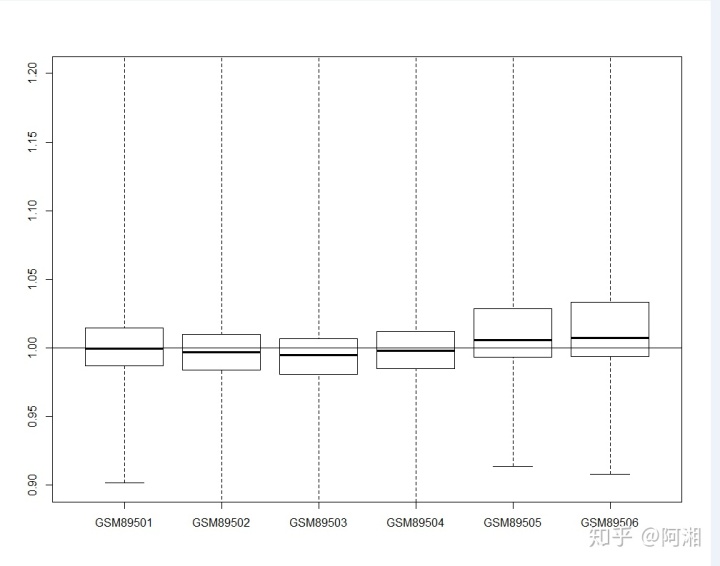
##3.2.5 使用RColorBrewer函数---RLE箱线图,绘制彩色的RLE图
library(RColorBrewer)
##载入一组颜色
n.cel <- length(cel.files)
colors<-brewer.pal(n = n.cel,name = "Set3")
##绘制RLE箱线图
pdf(file = "raw_data_RLE_Mbox_Rcolor.pdf", width = 12, height = 9)
Mbox(Pset,ylim=c(-1,1),col=colors,main="RLE",las=3)
dev.off()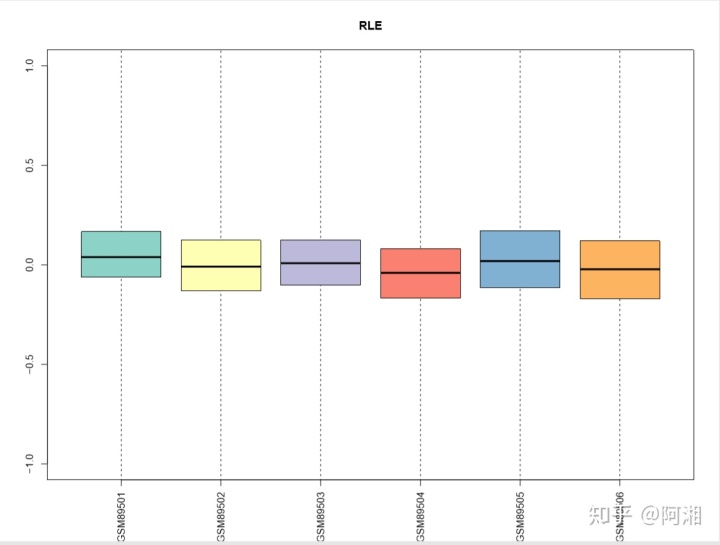
##3.2.6 使用彩虹色rainbow函数---RLE箱线图,rainbow是R的一个函数,用于产生彩虹色
n.cel <- length(cel.files)
colors<-rainbow(n.cel * 1.2)
##绘制RLE箱线图
pdf(file = "raw_data_RLE_Mbox_rainbow.pdf", width = 12, height = 9)
Mbox(Pset,ylim=c(-1,1),col=colors,main="RLE",las=3)
dev.off()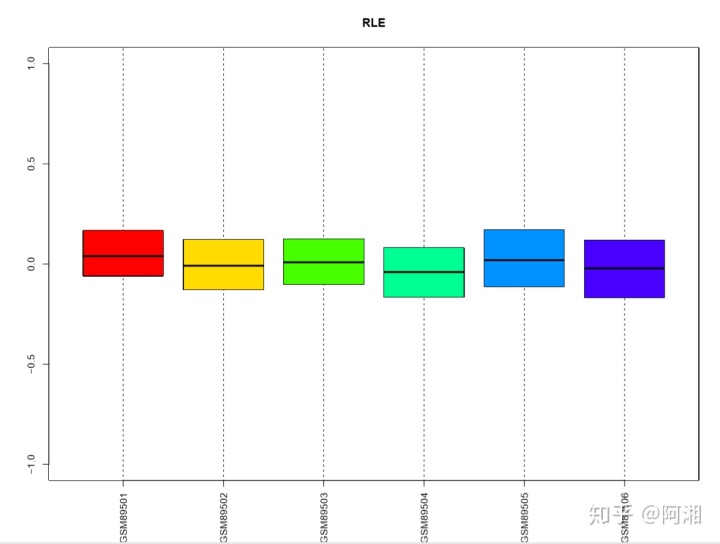
##3.2.7 使用彩虹色rainbow函数---NUSE箱线图,rainbow是R的一个函数,用于产生彩虹色
pdf(file = "raw_data_NUSE_boxplot_rainbow.pdf", width = 12, height = 9)
n.cel <- length(cel.files)
par(mfrow = c(1, 1))
par(mar = c(4, 4, 3, 0.5))
par(cex = 0.7)
if (n.cel > 40) par(cex = 0.5)
cols <- rainbow(n.cel * 1.2)
boxplot(Pset, col = cols, xlab = "Sample", ylab = "Log intensity")
dev.off()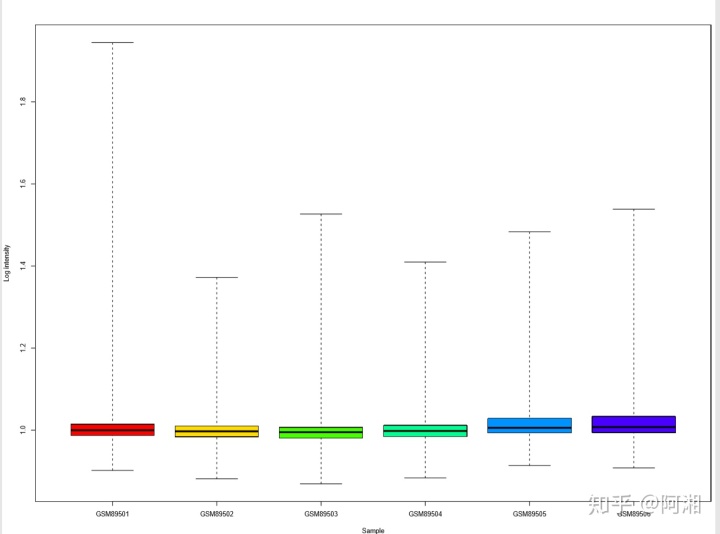
##3.2.8 使用 simpleaffy 包进行质量控制
library(simpleaffy)
library(ggplot2)
library(impute)
##获取质量分析报告
data.qc<-qc(data.raw)
##可视化质量分析报告
pdf(file = "raw_data_QC.pdf", width = 12, height = 9)
plot(data.qc)
dev.off()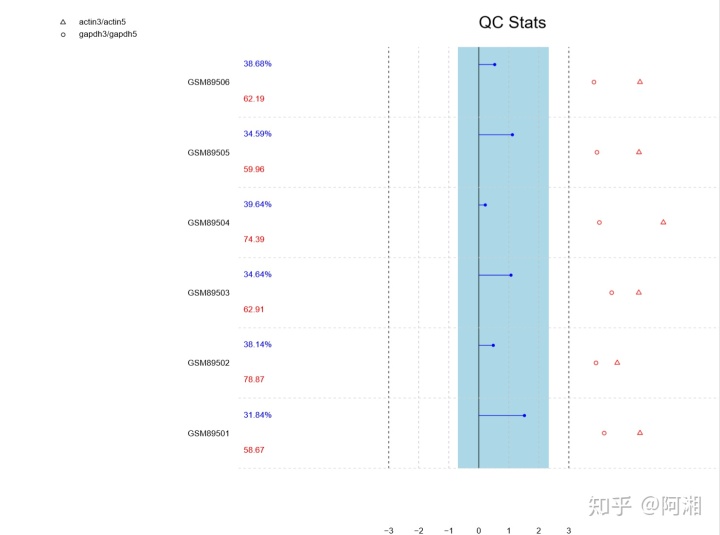
##3.2.9 RNA降解分析
#####rainbow颜色绘制RNA降解图
library(RColorBrewer)
##载入一组颜色
n.cel <- length(cel.files)
#cols<-brewer.pal(n = n.cel,name = "Set3")
#pallette.gray <- c(rep(gray(0:10/10), times = seq(1, 41, by = 4)))
#cols = pallette.gray
cols <- rainbow(n.cel * 1.2)
pdf(file = "RNAdeg_rainbow.pdf", width = 12, height = 9)
par(mfrow = c(1, 1))
par(mar = c(4, 4, 3, 0.5))
RNAdeg <- AffyRNAdeg(data.raw)
summaryAffyRNAdeg(RNAdeg)
plotAffyRNAdeg(RNAdeg, cols = cols)
legend("topleft", legend = sampleNames(data.raw), lty = 1, col = cols, box.col = "transparent",xpd = TRUE)
box()
dev.off()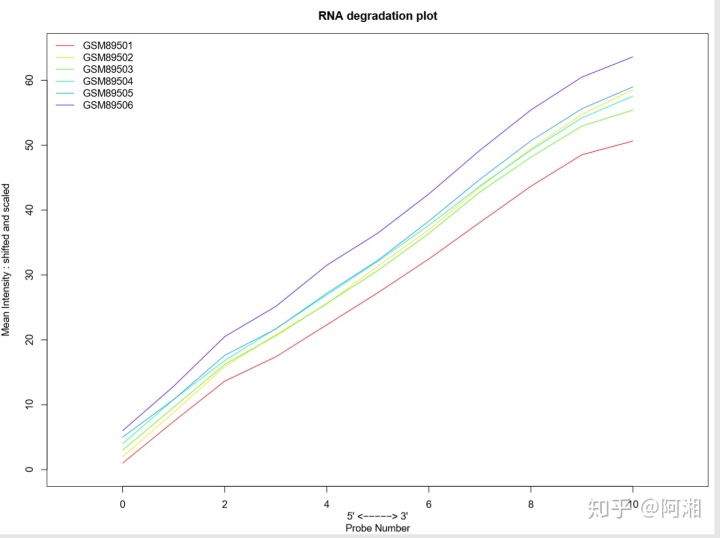
##3.2.10 使用 arrayQualityMetrics 包进行质量控制(一键生成上述所有图)
library(arrayQualityMetrics)
arrayQualityMetrics(expressionset = data.raw,
outdir = "fig",
force = TRUE,
do.logtransform = T)
dev.off()
#因该R包安装失败,故未能提供图##
##4.基因芯片数据预处理:背景校正,标准化,汇总
library(affy)
library(gcrma)
library(affyPLM)
library(RColorBrewer)
colors<-brewer.pal(12,"Set3")
#4.1 三合一函数
#eset.mas<-expresso(afbatch = CLLbatch,bgcorrect.method = "mas",
# normalize.method = "constant",pmcorrect.method ="mas",
# summary.method = "mas")##4.2 三种数据一体化处理函数任选择一个
##使用gcrma算法来预处理数据
data_raw_gcrma<-gcrma(data.raw)
##使用MAS5算法来预处理数据,mas5据说是expresso函数和mas方法的封装
##data.raw_mas5<-mas5(data.raw)
##使用rma算法来预处理数据,其背景处理方法为rma法,归一化处理使用分位数法,而汇总方法使用medianpolish
##data.raw_rma<-rma(data.raw)
sampleNames(data_raw_gcrma)#4.3 预处理后验证,此处以gcrma预处理为例
##4.3.1 绘制gcrma预处理后NUSE箱线图,使用彩虹色rainbow函数---NUSE箱线图,
library(affyPLM)
pdf(file = "data_raw_gcrma_NUSE_boxplot_rainbow.pdf", width = 12, height = 9)
n.cel <- length(sampleNames(data_raw_gcrma))
par(mfrow = c(1, 1))
par(mar = c(4, 4, 3, 0.5))
par(cex = 0.7)
if (n.cel > 40) par(cex = 0.5)
cols <- rainbow(n.cel * 1.2)
boxplot(data_raw_gcrma, col = cols, xlab = "Sample", ylab = "Log intensity")
dev.off()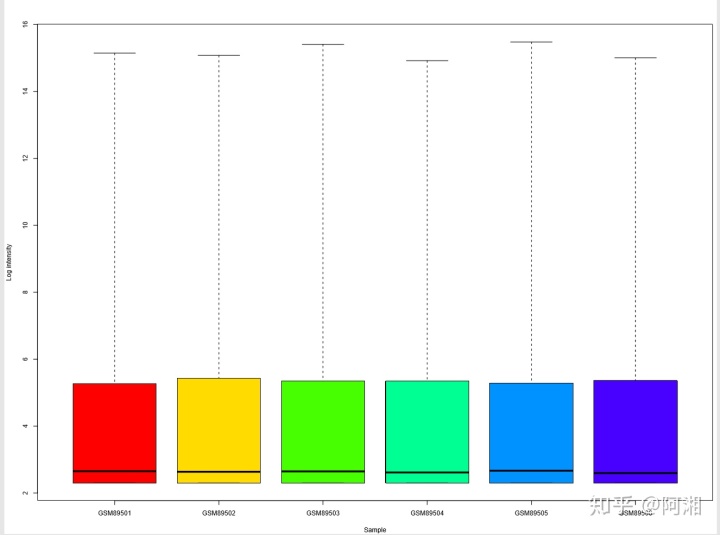
#4.3.2 直方图
##4.3.2.1 处理前hist
pdf(file="data_raw_hist.pdf",width=9,height=5)
par(mfrow = c(1,2))
cex1 = 0.9
colors=rainbow(n.cel * 1.2)
##原数据直方图
hist(data.raw,main="data_raw",col=colors)
legend('topright',rownames(pData(data.raw)),col=colors,lwd=1,inset=0.05,cex=0.5,ncol=3)
dev.off()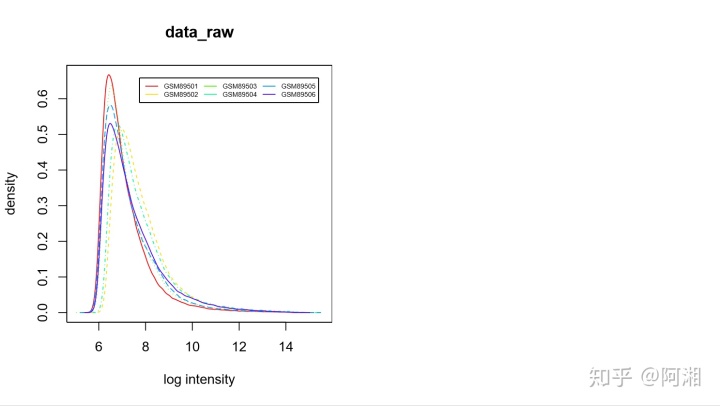
##4.3.2.2 处理后hist
pdf(file="data_raw_gcrma_hist.pdf",width=9,height=5)
par(mfrow = c(1,2))
cex1 = 0.9
colors=rainbow(n.cel * 1.2)
hist(data_raw_gcrma,main="gcrma",col=colors)
legend('topright',rownames(pData(data_raw_gcrma)),col=colors,lwd=1,inset=0.05,cex=0.5,ncol=3)
dev.off()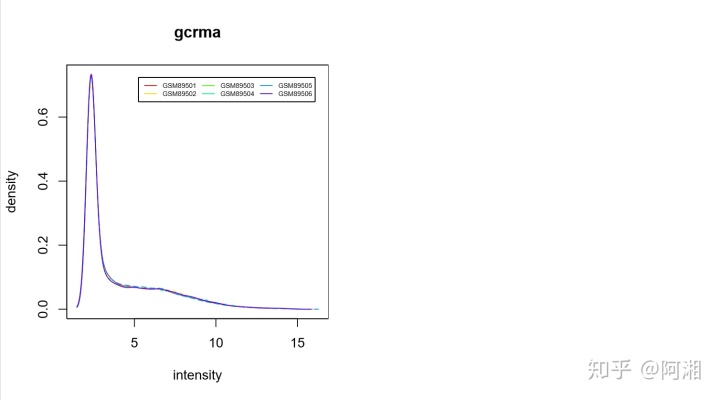
##4.3.2.3 预处理后可进行聚类分析,发现异类样本
eset<-exprs(data_raw_gcrma)##提取基因表达矩阵
pearson_cor<-cor(eset)##计算样品两两之间的Pearson相关系数
dist.lower<-as.dist(1-pearson_cor)##得到pearson距离的下三角矩阵
pdf(file = "eset_hclust_plot.pdf", width = 12, height = 9)
##聚类分析
hc<-hclust(dist.lower,"ave")
plot(hc,hang=-1)
dev.off()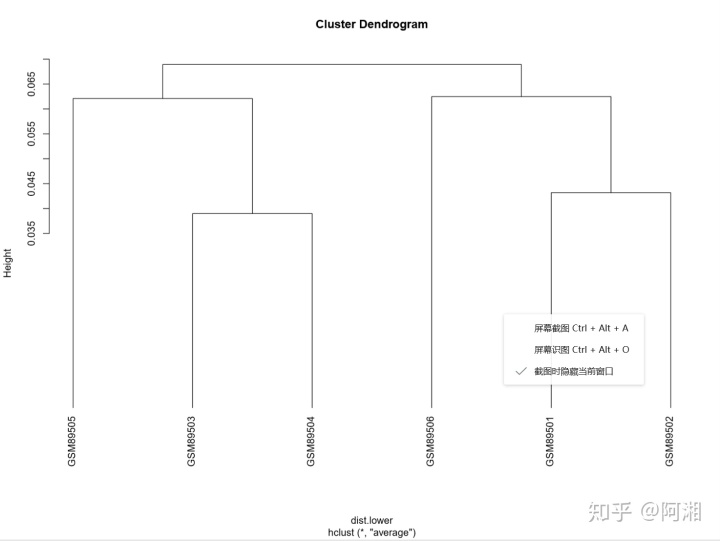
##4.3.2.4 PCA分析--没做出来,尴尬
library(Biobase)
library(BiocGenerics)
eset<-exprs(data_raw_gcrma)##提取基因表达矩阵
#sampleNames<-sub(pattern = ".CEL",replacement = "",colnames(eset)) ##PCA分析
sampleNames=sampleNames(data_raw_gcrma)
phenotype<-group_file
phenotype
groups<-factor(phenotype[,"disease"])#提取实验条件信息
pdf(file = "PCA_plot.pdf", width = 12, height = 9)
plotPCA(eset,addtext=sampleNames,groups=groups,groupnames=levels(groups))
dev.off()
****#综合分析上述结果,决定芯片是否使用
##5 构建余下样品的基因表达矩阵,用于后续分析
##假如此处GSM89501异常可能性大,则剔除
data<-data_raw_gcrma[,-match("GSM89501",rownames(pData(data_raw_gcrma)))]
dataExpr0=exprs(data)
colnames(dataExpr0)#如果无数据需要删除,则直接进入下一步
##获取样本各基因表达矩阵
data_exp=exprs(data_raw_gcrma)
save(dataexp,files="GSE**_exp.RData")
write.csv(dataexp,file="GSE**_exp.csv",quote=F)
#####limma包配对续差异基因分析
学习过程的就是分享的过程,分享的过程也是交流的过程,交流的过程就是进步的过程。