我们演示的数据集是GSE19136,样本可分为三组(对照组,支架组,紫杉醇组),具体实验设计情况请查看链接。本篇文章主要对对照组和支架组 做差异分析。分析流程主要包括基于limma包的差异分析、pheatmap包的热图和ggplot2的火山图(包括对基因的批量标记)。
1. 差异分析
- 数据下载
setwd("D:RwjhGSE19136")
library(GEOquery) #用GEOquery包获取GEO数据
eSet<-getGEO('GSE19136',destdir='./',getGPL=F) #下载数据,构建eSet对象
eset<-exprs(eSet[[1]])[,c(1,2,4,5,7,8,10,11)] #提取基因表达矩阵
metadata<-pData(eSet[[1]]) #可以查看芯片的描述性信息,为后面分组做准备
x<-metadata$title[c(1,2,4,5,7,8,10,11)] #查看芯片的标题,可以选择用于分组的字段
group_list<-factor(unlist(lapply(x,function(x) strsplit(as.character(x),"_")[[1]][5]))) #我们发现取第5个字段作为分组信息最好- ID转换
gpl <- getGEO('GPL570', destdir=".") #下载并获取GSE19136对应平台的注释信息
anno1<-Table(gpl)[,c(1,11)] #提取探针ID,gene symbol
eset<-as.data.frame(eset) #矩阵转换成数据框,便于后面添加非数值型的列
eset$ID <- rownames(eset) #添加一列(基因名)
merg<-merge(eset,anno1,by="ID") #合并表达矩阵和基因名,通过探针号索引
y<-merg$`Gene Symbol`
gene<-unlist(lapply(y,function(y) strsplit(as.character(y)," /// ")[[1]][1]))
merg$gene <- gene #一个探针对应多个基因的时候,取第一个基因
aggr<-aggregate(merg[,2:9],by=list(merg$gene),mean) #多个探针对应一个基因的时候,取平均值- 差异分析
eset <- aggr[,2:9]
rownames(eset)<-aggr[,1] #得到一个行名为unique的gene symbol矩阵
design<-model.matrix(~-1+group_list) #构建实验设计矩阵
library(limma)
contrast.matrix<-makeContrasts(contrasts = "group_listbare-group_listcontrol", levels = design) #构建对比模型,比较两个实验条件下表达数据,根据上面的group_list修改bare和control
fit <- lmFit(log2(eset),design) #线性模型拟合
fit1 <- contrasts.fit(fit, contrast.matrix) #根据对比模型进行差值计算
fit2 <- eBayes(fit1) #贝叶斯检验
tempOutput = topTable(fit2, coef="group_listbare-group_listcontrol", n=nrow(fit2),lfc=log2(1)) #生成所有基因的检验结果报表
dif<-tempOutput[tempOutput[,"P.Value"]<0.05,] #根据P值筛选全部差异表达基因(图)
dim(dif)
head(dif)
write.csv(dif,file = "dif.csv",quote = F)2. 画热图
rt <- read.table("dif.csv",header = T,sep = ",",stringsAsFactors = F)
heat<-eset[rownames(eset) %in% c(head(rownames(subset(dif,dif$logFC>0)),15),head(rownames(subset(dif,dif$logFC<0)),15)),] #对前15个上调基因和下调基因做热图
library(pheatmap)
x <- t(scale(t(heat))) #事实证明用这个做归一化,效果最好
pheatmap(x)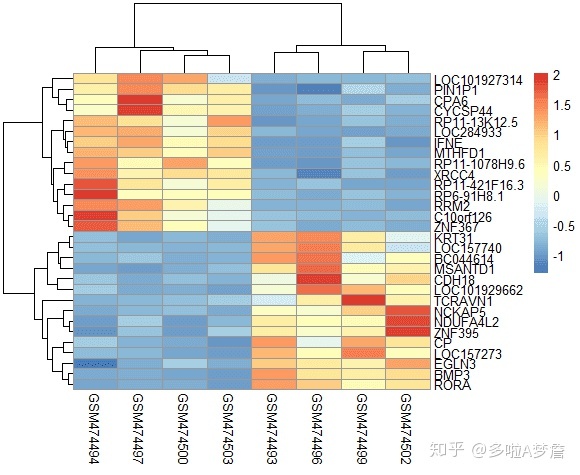
3. 火山图
##################### 火山图 #################################
tempOutput1 = topTable(fit2, coef="group_listbare-group_listcontrol", n=nrow(fit2),lfc=log2(1)) #生成所有基因的检验结果报表
data<-tempOutput1[tempOutput1[,"P.Value"]<=1,]
data$gene<-rownames(data)
data$sig[data$P.Value>=0.05 | abs(data$logFC) < 1] <- "black"
data$sig[data$P.Value<0.05 & data$logFC >= 1] <- "red"
data$sig[data$P.Value<0.05 & data$logFC <= -1] <- "green"
library(ggplot2)
library(ggrepel)
library(dplyr)
ggplot(data=data, aes(x=logFC, y =-log10(P.Value),color =sig)) +
geom_point() +theme_set(theme_bw())+theme(panel.grid=element_blank(),strip.text = element_blank())+
scale_color_manual(values = c("black","green","red"))+
geom_hline(yintercept = -log10(0.05),lty=4,lwd=0.6,alpha=0.8)+
geom_vline(xintercept = c(log2(2),-log2(2)),lty=4,lwd=0.6,alpha=0.8)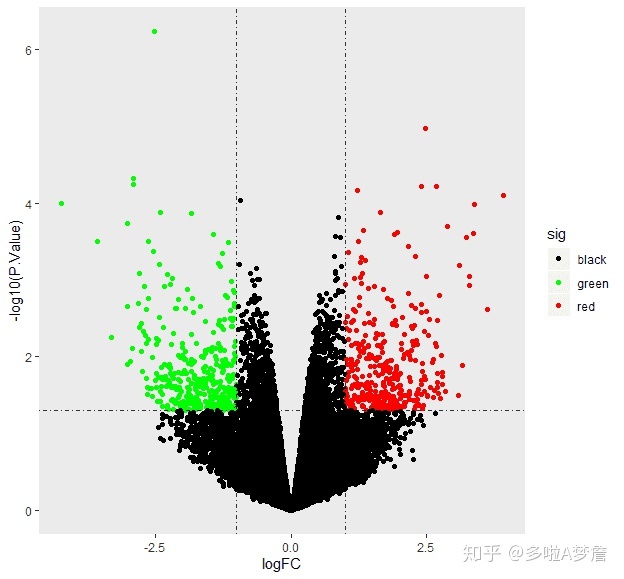
4. 基因标记的火山图
input <- data
Gene<-as.data.frame(row.names(heat))
volc = ggplot(input, aes(logFC, -log10(P.Value))) +
geom_point(aes(col=sig)) + scale_color_manual(values=c("black","green", "red")) +
ggtitle("Your title here")+geom_point(data=input[input$gene %in% Gene[,1],], aes(logFC, -log10(P.Value)), colour="blue", size=2) +
geom_hline(yintercept = -log10(0.05),lty=4,lwd=0.6,alpha=0.8)+
geom_vline(xintercept = c(log2(2),-log2(2)),lty=4,lwd=0.6,alpha=0.8)
volc+geom_text_repel(data=input[input$gene %in% Gene[,1],], aes(label=gene))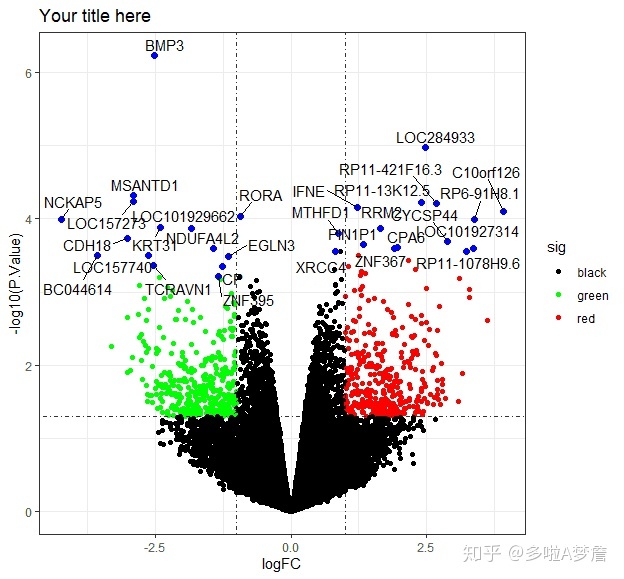
5. GO和KEGG
library(clusterProfiler)
library(pathview)
rt <- read.table("dif.csv",header = T,sep = ",",stringsAsFactors = F)
colnames(rt)[1] <- "SYMBOL"
GeneSymbol <- rt$SYMBOL
gene.symbol.eg<- id2eg(ids=GeneSymbol, category='SYMBOL', org='Hs',na.rm = F) #如果是小鼠,就是Mm
merg<-merge(gene.symbol.eg,rt,by="SYMBOL")
mer <- subset(merg,merg$ENTREZID != "NA")
geneFC <- mer$logFC
gene <- mer$ENTREZID
names(geneFC) <- gene
kkd <- enrichGO(gene = gene,"org.Hs.eg.db",ont = "BP",qvalueCutoff = 0.05) #如果是小鼠,就是Mm
write.table(kkd@result,file = "GO.xls",sep = "t",quote = F,row.names = F)
barplot(kkd,drop=T,showCategory = 12)
cnetplot(kkd,categorySize = "geneNum",foldChange = geneFC)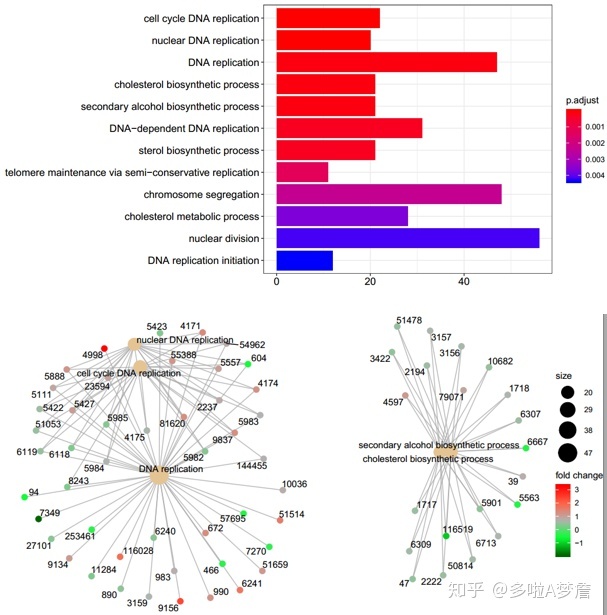
kk <- enrichKEGG(gene = gene,organism = "human",qvalueCutoff = 0.05) #如果是小鼠,就是mouse
write.table(kk@result,file = "KEGG.xls",sep = "t",quote = F,row.names = F)
barplot(kk,drop=T,showCategory = 12)
cnetplot(kk,categorySize = "geneNum",foldChange = geneFC)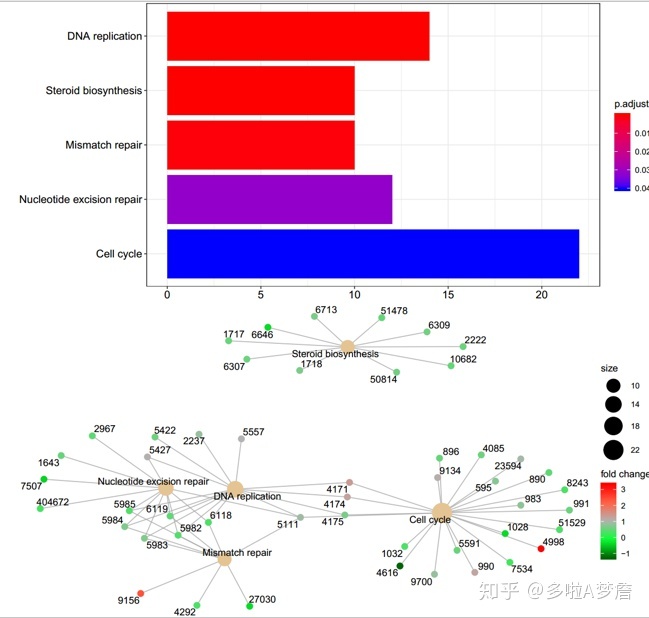
setwd("./pathview") #新建pathview文件夹,存放KEGG关系图
keggxls<-read.table("KEGG.xls",sep = "t",header = T)
keggxls<-subset(keggxls,keggxls$p.adjust<0.05)
for (i in keggxls$ID){
pv.out <- pathview(gene.data = geneFC, pathway.id = i, species = "hsa", out.suffix = "pathview") #如果是小鼠mmu,看GSE70410
}6. PPI
ppi<-read.table("string_interactions0.5.tsv",header = T,sep = "t",stringsAsFactors = F)
ppi_attr1 <- rt[rt$SYMBOL %in% unique(c(ppi[,1],ppi[,2])),] # rt即是dif
ppi_attr <- merge(ppi_attr1,as.data.frame(table(c(ppi[,1],ppi[,2]))),by.x="SYMBOL",by.y="Var1")
write.csv(ppi_attr,file = "ppi_attrabute.csv",quote = F,row.names = F)
write.table(ppi_attr,file = "ppi_attrabute.txt",quote = F,sep = "t",row.names = F)如果觉得好,请给我一点动力!
版权声明:本文为weixin_31968223原创文章,遵循CC 4.0 BY-SA版权协议,转载请附上原文出处链接和本声明。