GO、KEGG分析
1、基本分析
library(clusterProfiler)
library(org.Hs.eg.db)
options(clusterProfiler.download.method = "wininet") #不加上这句,KEGG分析会失败
GO_BP = enrichGO(gene = idTable$ENTREZID,
OrgDb = org.Hs.eg.db,
keyType = "ENTREZID",
ont = "BP",
pvalueCutoff = 0.9,qvalueCutoff = 0.9) #只输出在p和q的阈值内的富集通路,可设大一些
KEGG <- enrichKEGG(gene = idTable$ENTREZID,
organism = 'hsa',
keyType = 'kegg',
pvalueCutoff = 0.9,qvalueCutoff = 0.9)
GO_BP.df = as.data.frame(GO_BP) #可输出为df格式便于浏览
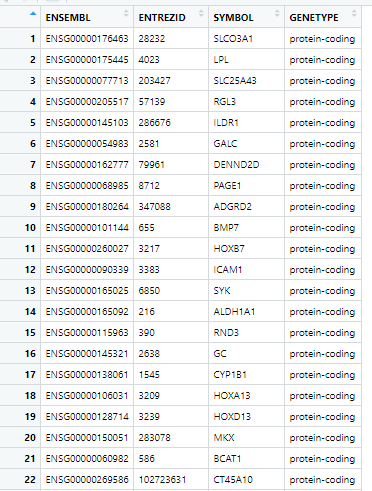
KEGG.df = as.data.frame(KEGG)- GO和KEGG分析需要输入基因ID向量,输入的基因ID的类型一般选择EntrezID
- 不同物种有不同的基因注释包,链接:Bioconductor - BiocViews
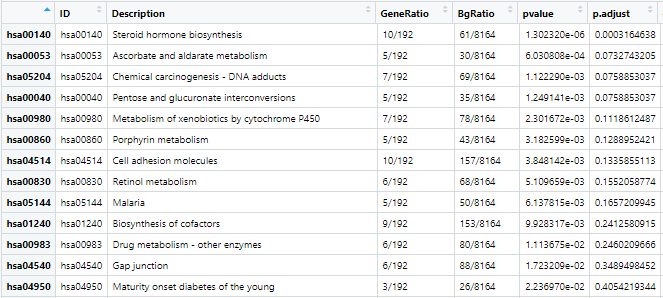
- 可将富集结果输出为数据框便于浏览,如 GO_BP.df,且df按 pvalue 进行排序:
2、可视化
dotplot(GO_BP,
showCategory=10, #展示通路数
title="GO_BP")
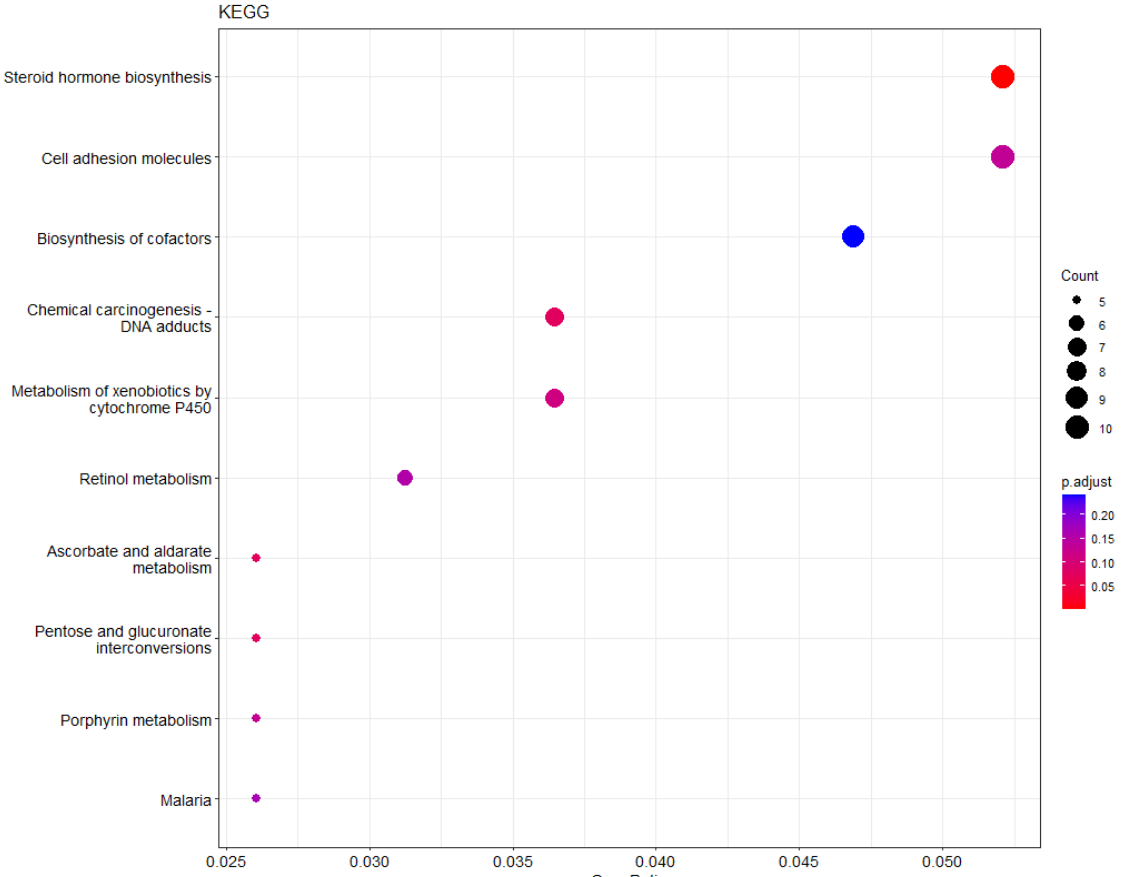
dotplot(KEGG,
showCategory=10,
title="KEGG")
版权声明:本文为weixin_59289660原创文章,遵循CC 4.0 BY-SA版权协议,转载请附上原文出处链接和本声明。