一、KEGG
1、http://www.genome.jp/kegg/,京都基因与基因组百科全书;某基因、转录本参与的具体生物学通路情况
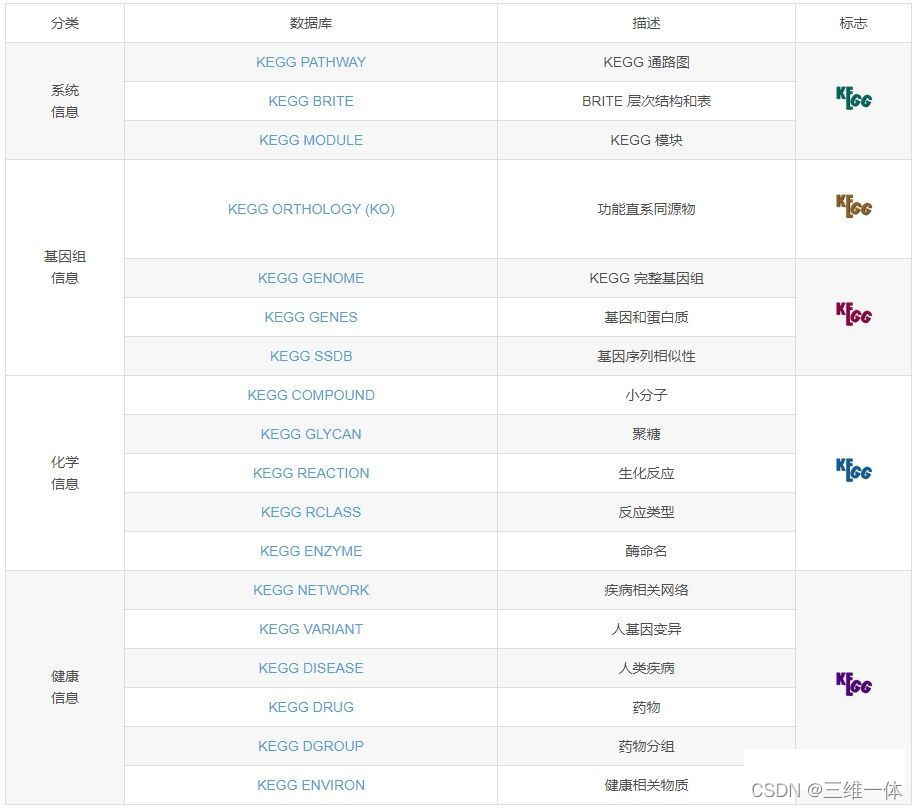
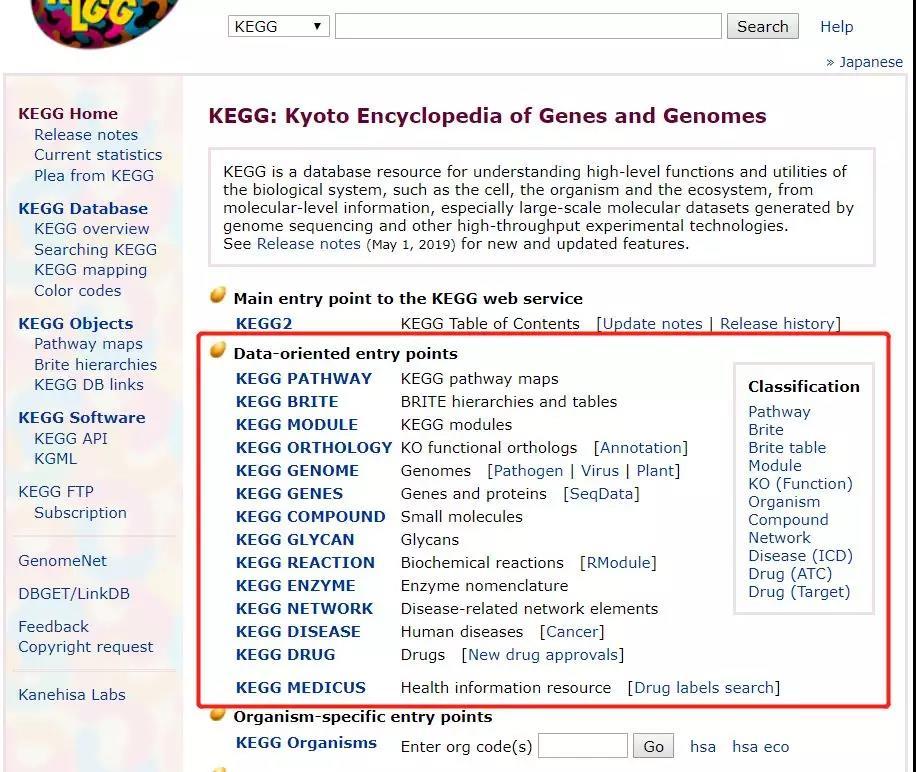
2、分类
3、KEGG详细解读:
- 各功能模块描述
一文快速读懂 KEGG 数据库与通路图 - 知乎 (zhihu.com)![]() https://zhuanlan.zhihu.com/p/96008506
https://zhuanlan.zhihu.com/p/96008506
- 通路查询
如何从KEGG数据库找到想要的通路? - 知乎 (zhihu.com)![]() https://zhuanlan.zhihu.com/p/401700324
https://zhuanlan.zhihu.com/p/401700324
4、运用:基因集GSEA分析,差异基因KEGG功能富集分析
二、GO
1、GO/Gene Ontology 基因本体论, http://www.geneontology.org,由基因本体论联合会建立,对基因、蛋白功能进行统一限定和描述。通过GO富集分析可粗略了解基因富集在哪些生物学功能、途径或细胞定位。
2、分类:
BP:Biological Process,生物过程
MF:Molecular Function,分子功能
CC:Cellular Component,细胞组分
三、Hallmark geneset 效应特征基因集:该类别包含多个已知基因集构成的超基因集,每个类别的基因集都对应多个基础的其他类别的基因集。
四、运用:基因集GSEA、GSVA,差异基因功能富集分析,单细胞佐证亚群分群
上述基因集都在MSigDB数据库中,也是进行GSEA的必用基因集数据库
- GSEA官网:GSEA | Login (gsea-msigdb.org)
 http://www.gsea-msigdb.org/gsea/login.jsp;jsessionid=F4F18E65C6B7F24A66CF1B9A3393165D
http://www.gsea-msigdb.org/gsea/login.jsp;jsessionid=F4F18E65C6B7F24A66CF1B9A3393165D - MSigDB数据库介绍
GSEA分析是个什么鬼?(上) (360doc.com)![]() http://www.360doc.com/content/16/1026/18/19913717_601568937.shtml六、GSEA基因集富集分析
http://www.360doc.com/content/16/1026/18/19913717_601568937.shtml六、GSEA基因集富集分析
1、富集分析及原理:生物过程通常由一组基因参与,若生物过程发生异常,则相关基因组成与这一过程相关的基因集合。富集分析比较基因集在某个功能上相比随机水平是否发生过出现现象(over-presentation),显示此生物功能大体上下调趋势,其无法显示基因上下调趋势
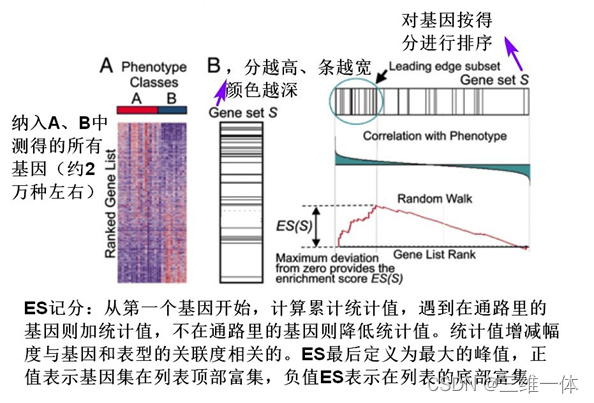
2、与传统GO/KEGG富集分析比较:GO/KEGG富集先筛差异基因,再判断差异基因在哪些通路富集,涉及阈值设定,存在一定主观性且只能用于表达变化较大的基因,而GSEA不局限于差异基因,是从基因集的富集角度出发
3、R实现
######################################################NGS GSEA
##整理数据
select.FPKM <- (res2$baseMean > 10) #初筛样本表达基因
rownames(res2)
degs.list=as.character(res2$symbol)[select.FPKM] #提取res2 基因名列符合筛选条件的基因名
res3=distinct(res2,symbol,.keep_all = T) #去重
rownames(res3)=res3$symbol #行名变为基因名
f1=res3[degs.list,] #提取筛选后基因名的行
exp.fc=f1[,c(2,4)] #提取基因名列及FC值列
##id转化并按FC降序排列
expm.id <- bitr(exp.fc$symbol, fromType = "SYMBOL", toType = "ENTREZID", OrgDb = "org.Hs.eg.db") #ID转换
exp.fc.id <- merge(exp.fc, expm.id,by.x="symbol", by.y="SYMBOL", all=F) #转换后的ID与FC文件融合
exp.fc.id=na.omit(exp.fc.id) #去空
exp.fc.id.sorted <- exp.fc.id[order(exp.fc.id$log2FoldChange, decreasing = T),] #降序排列FC
#exp.fc.id.sorted <-arrange(exp.fc.id,desc(log2FoldChange))
id.fc <- exp.fc.id.sorted$log2FoldChange #提取FC列
names(id.fc) <- exp.fc.id.sorted$ENTREZID #提取ENTREZID并于FC对应
##GSEA分析
gsea <- gseKEGG(id.fc, organism = "hsa",pvalueCutoff = 0.05)
A=gsea@result #查看富集结果,NES>1.5,p<0.05的显著富集
#可视化
gseaplot2(gsea,geneSetID = 1,title = "Hippo signaling pathway",color = "green",base_size = 10,pvalue_table = T) #画第一个通路的富集图,可输入通路ID“”
gseaplot2(gsea, geneSetID = c(2,3))
###使用msigdbr包内的通路数据库进行富集分析:更新较慢,最好使用上述官网数据库
##数据整理,同上
select.FPKM <- (res2$baseMean > 10) #基因初筛、提取基因名
degs.list=as.character(res2$symbol)[select.FPKM]
res3=distinct(res2,symbol,.keep_all = T) #去重,整理数据
rownames(res3)=res3$symbol
f1=res3[degs.list,]
exp.fc=f1[,c(2,4)]
deg = exp.fc$log2FoldChange
names(deg) = exp.fc$symbol
deg= sort(deg,decreasing = T) #order、sort针对不同数据类型
h.kegg= msigdbr(species ="Homo sapiens")
length(unique(h.kegg$gs_exact_source)) #unique():返回参数中所有的不同值,并按从小到大排序
h.kegg= h.kegg[,c(3,4)]
gsea <- GSEA(deg,pvalueCutoff = 0.5, TERM2GENE =h.kegg) #GSEA()函数富集分析
gsea1=gsea@result
gseaplot2(gsea, geneSetID = 6, title = gsea$Description[6])
write.table(gsea1,"gsea.res.txt",sep = "\t",col.names = NA)
############################################################### ★★单细胞 fGSEA 快速富集
# fGSEA的预选基因集可以自己设定,一般使用MSigdb数据库
# fgsea()函数需要一个基因集列表以及对应值,主要是基因名和AUC(ROC曲线下面积,准确性越高,AUC值越大),基因集中的基因名要与数据集(pathway)中的基因名相对应。
# plotEnrichment()函数用于GSEA图的绘制
library(msigdbr)
library(fgsea)
##获取小鼠富集通路数据集
msigdbr_species() #查看各物种富集通路数据库
m_df<- msigdbr(species = "Mus musculus",
category = "C2", #GSEA官网查看类别、亚类别
subcategory = "KEGG")
fgsea_sets<- m_df %>% #切割分组构建通路基因集list
split(x = .$gene_symbol, #分组对象
f = .$gs_name) #切割依据
##提取基因差异倍数并重命名
genelist=deg$avg_log2FC
names(genelist)= rownames(deg)
genelist=sort(genelist,decreasing = T) #降序排列
##富集分析及可视化
fgseaRes<- fgsea(fgsea_sets, #通路基因集
stats = genelist ) #差异变化倍数
fp=ggplot(fgseaRes %>%
filter(padj < 0.005) %>% #过滤筛选
head(n= 20),
aes(reorder(pathway, NES), NES)) + #NES:标准化ES,<1.5下调
geom_col(aes(fill= NES < 1.5)) +
coord_flip() +
labs(x="Pathway", y="Normalized Enrichment Score",
title="KEGG pathways NES from GSEA") +
theme_minimal() 4、结果分析
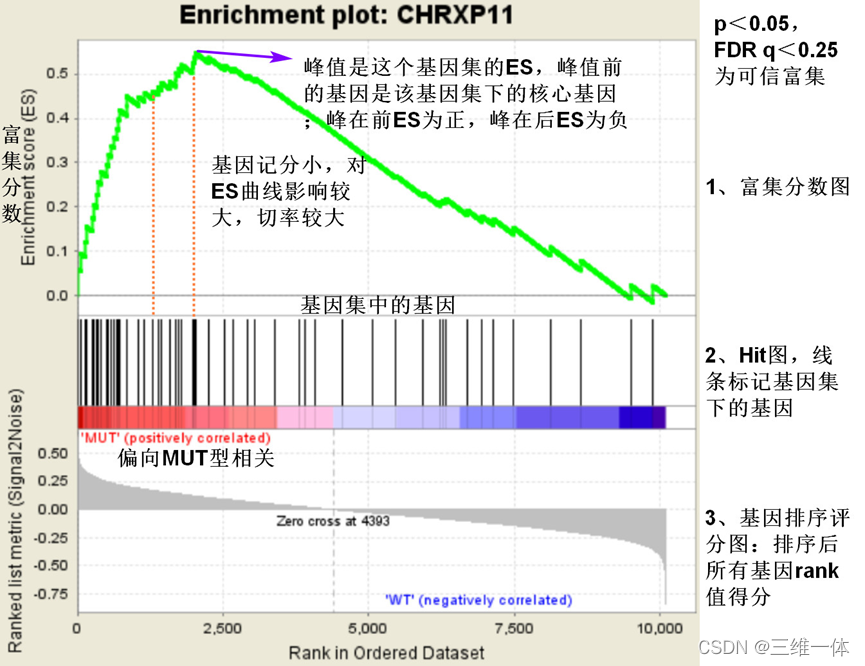
SIZE:表示基因集里的基因数量
RANK AT AMX:ES值对应的通路基因排名
ES(enrichment score):富集分数
NES(normalized enrichment score):表示校正后的富集分数
NOM p-val (nominal p value ): 名义P值
FDR q-val(false discovery rate):错误发现率≈p val
FWER p-val:用bonferonni校正后的P值
Leading-edge subset:对富集贡献最大的基因成员,即领头亚集
- 通常认为|NES|>1,p-val<0.05,FDR q-val<0.25的通路下的基因集是有意义的;NES绝对值越大、FDR值越小说明结果可信度越高。当p-val很小、FDR值很大时,说明它的富集并不显著,可能是样本量较少、杂交信号微弱、选择的功能基因集并未较好的反映样本
- ES折线图:纵轴值全部大于0则这个基因集在MUT组高表达峰值左侧为核心基团,若为负数峰值右侧为核心基因
- Rank值类似标准化log2FC值,是所有感兴趣基因的值
- 注意:
调整分析的基因、通路数据库来源、参数调整可以调整预期结果;不同数据库通路结果可能不完全一致,但总体趋势差不多,可用多数据库处理验证结果
人类基因名:全部大写;鼠类基因名:开头大写,后面小写
GSEA数据库中都是人的基因数据,NCBI gene数据库查找相关基因具体信息及人鼠差异
七、ssGSEA 单样本基因集富集分析
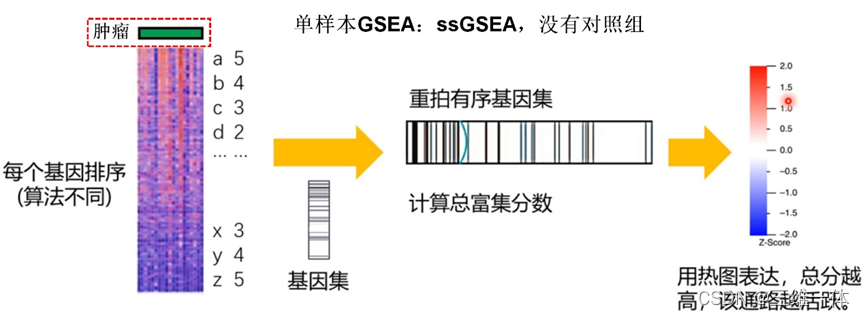

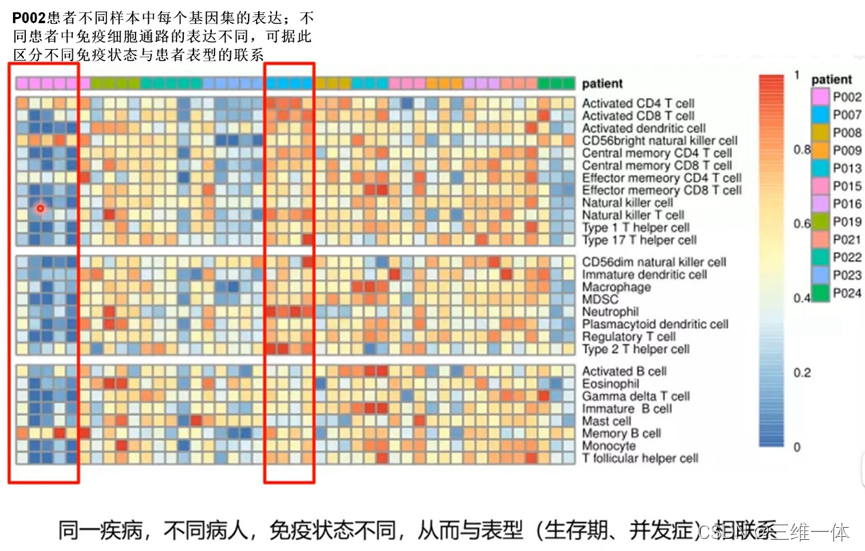
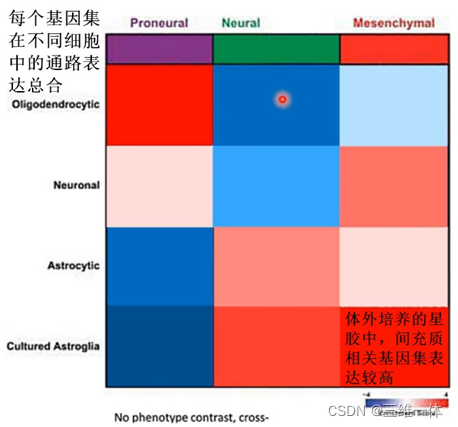
八、GSVA
1、基因集变异分析 GSVA是其中一个GSEA算法。也可以一开始从基因表达量和多个通路信息出发,根据通路活性变化情况,对样本进行无监督分类。
2、GSEA、GSVA、ssGSEA、GO/KEGG富集分析区别及联系
- 从输入的表达矩阵入手
- GSEA输入矩阵:差异分析后得到的矩阵:一列基因名/ENTREZ + logFC值(需按照logFC值从大到小排列)
- GSVA输入矩阵:列是样本、行是基因、单元格内是表达量【没有差异分析!】
- ssGSEA:长得像GSEA,其实是GSVA的好兄弟,因为单样本无法进行差异分析就无法进行GSEA
- GO/KEGG输入矩阵:只用提供基因名
- 分析方法
- GSEA:基于如下假设: 一个基因集中的基因在表达谱中所处的Rank越极端、基因间距越短(Rank相近),该基因集越显著
- GSVA:
- 如果某个基因存在于某个通路,那就给它“一分”,不在就扣它“一分”,这样就能计算得到Enrichment Score(ES)
- 这样GSVA后的表达矩阵变成了:列是样本,行是通路,单元格是ES
- ssGSEA:只有一个样本,计算方法≈GSVA
3、R实现
library(TCGAbiolinks)
library(GSVA)
library(GSEABase)
# 获取表达矩阵
# 自定义函数用于下载TCGA各癌肿表达矩阵为例
get_count <- function(cancer, n = 10) {
query <- GDCquery(
project = cancer,
data.category = "Gene expression",
data.type = "Gene expression quantification",
platform = "Illumina HiSeq",
file.type = "results",
sample.type = c("Primary Tumor"),
legacy = TRUE
)
# 选择 n 个样本
query$results[[1]] <- query$results[[1]][1:n,]
GDCdownload(query)
#获取 read count
exp.count <- GDCprepare(
query,
summarizedExperiment = TRUE,
)
return(exp.count)
}
luad.count <- get_count("TCGA-LUAD")
lusc.count <- get_count("TCGA-LUSC")
dataPrep_luad <- TCGAanalyze_Preprocessing(
object = luad.count,
cor.cut = 0.6,
datatype = "raw_count"
)
dataPrep_lusc <- TCGAanalyze_Preprocessing(
object = lusc.count,
cor.cut = 0.6,
datatype = "raw_count"
)
# 合并数据并使用 gcContent 方法进行标准化
dataNorm <- TCGAanalyze_Normalization(
tabDF = cbind(dataPrep_luad, dataPrep_lusc),
geneInfo = TCGAbiolinks::geneInfo,
method = "gcContent"
)
# 分位数过滤
dataFilt <- TCGAanalyze_Filtering(
tabDF = dataNorm,
method = "quantile",
qnt.cut = 0.25
)
# 将数据拆分
luad.exp <- subset(dataFilt, select = luad.count$barcode)
lusc.exp <- subset(dataFilt, select = lusc.count$barcode)
# 读取从 GSEA 官网下载的通路数据
c2gmt <- getGmt("/Volumes/Seagate/c2.cp.v7.4.symbols.gmt")
# 筛选出常用的这三个数据库中的通路
gene.set <- c2gmt[grep("^KEGG|REACTOME|BIOCARTA", names(c2gmt)),]
# gsva 分析,read counts 使用泊松分布,通路至少包含 10 个基因;连续型的数据(如微阵列荧光/RNA-seq/log-CPMs/log-RPKMs/log-TPMs)用Gaussian,整数型的数据用 Poisson
gs.exp <- gsva(dataFilt, gene.set, kcdf = "Poisson", min.sz = 10)
# limma差异分析
# 上述GSVA分析把行名从基因名变成了PATHWAY名,便可以分析差异通路了
DEA.gs <- TCGAanalyze_DEA(
mat1 = gs.exp[, colnames(luad.exp)],
mat2 = gs.exp[, colnames(lusc.exp)],
metadata = FALSE,
pipeline = "limma",
Cond1type = "LUAD",
Cond2type = "LUSC",
fdr.cut = 0.05,
logFC.cut = 0.5,
)
## ssGSEA
res.ssgsea <- gsva(dataFilt, gene.set, method = "ssgsea", kcdf = "Poisson", min.sz = 10)
# 热图
# 设置分组:1是LUAD:2是LUSC
annotation_col <- data.frame(sample = rep(1:2, each = 10))
rownames(annotation_col) <- colnames(gs.exp)
# png("/Users/apple/Desktop/R-Scripts/GSEA/DEA.gs.hm.png",width = 1000,height = 1000)
pdf("/Users/apple/Desktop/R-Scripts/GSEA/DEA.gs.hm.pdf")
pheatmap(
gs.exp[rownames(DEA.gs),],
show_colnames = F,
# 不展示行名
cluster_rows = F,
# 不对行聚类
cluster_cols = F,
# 不对列聚类
annotation_col = annotation_col,
# 加注释
cellwidth = 5,
cellheight = 5,
# 设置单元格的宽度和高度
fontsize = 5
)
dev.off()